Summary
Thrombocytopenia means a platelet count below 150,000/mm³. Clinically significant bleeding (mucocutaneous bleeding, petechiae, purpura) usually appears when platelets drop below 50,000/mm³, and spontaneous severe bleeding when below 10,000–20,000/mm³.
This lesson focuses on the four most exam-tested causes:
- ITP – Immune Thrombocytopenic Purpura: autoantibodies destroy platelets. Isolated low platelets, otherwise normal labs.
- TTP – Thrombotic Thrombocytopenic Purpura: ADAMTS13 deficiency → large vWF multimers → microthrombi. Pentad: fever, neuro symptoms, renal failure, MAHA, thrombocytopenia.
- HUS – Hemolytic Uremic Syndrome: Shiga toxin from E. coli O157:H7. Triad: MAHA, thrombocytopenia, acute kidney injury. Usually in children after bloody diarrhea.
- HIT – Heparin-Induced Thrombocytopenia: IgG antibodies against heparin-PF4 complex. Paradoxical thrombosis, not bleeding, 5–10 days after heparin exposure.
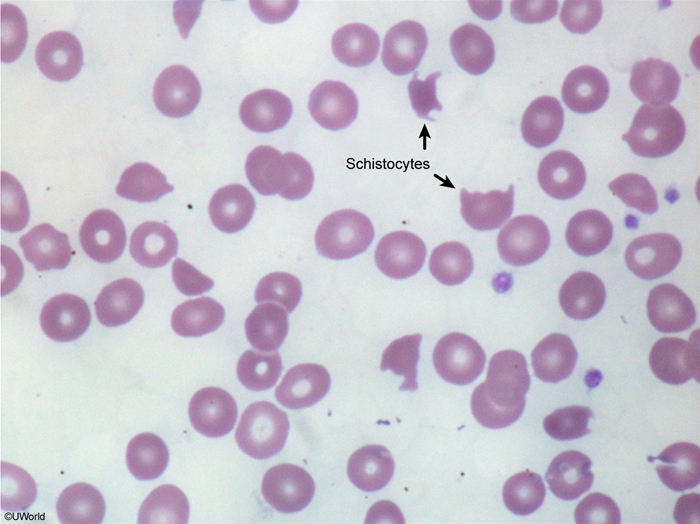
The single most important question to ask is: "Is this isolated thrombocytopenia, or is there also hemolysis (schistocytes on smear)?" Isolated → think ITP. With schistocytes → think TTP/HUS/DIC.
Approach to Thrombocytopenia
When you see a low platelet count, work through three quick questions:
- Is it real? Rule out pseudothrombocytopenia (EDTA-induced platelet clumping). Repeat in a citrate tube.
- Are other cell lines affected?
- Isolated low platelets → ITP, drug-induced, HIT.
- Low platelets + anemia + schistocytes → TTP, HUS, DIC (microangiopathic hemolytic anemia, MAHA).
- Pancytopenia → bone marrow problem (aplastic anemia, leukemia, B12 deficiency).
- Are coagulation tests normal or abnormal?
- Normal PT/PTT/fibrinogen → ITP, TTP, HUS, HIT.
- Prolonged PT/PTT + low fibrinogen → DIC (consumptive coagulopathy).
Clinical signs of platelet deficiency are mucocutaneous: petechiae, purpura, epistaxis, gum bleeding, menorrhagia, and easy bruising.
Immune Thrombocytopenic Purpura (ITP)
Definition: Acquired autoimmune disorder in which IgG autoantibodies against platelet surface glycoproteins (GpIIb/IIIa) cause splenic destruction of platelets.
Epidemiology & triggers
- Children: acute, self-limited; follows a viral infection (URI, EBV, varicella) by 1–4 weeks.
- Adults: chronic; women > men. May be associated with SLE, HIV, HCV, CLL, or H. pylori.
Clinical features
- Sudden mucocutaneous bleeding: petechiae, purpura, epistaxis, gingival bleeding, menorrhagia.
- Patient otherwise looks well – no fever, no organomegaly, no neurologic signs.
- Splenomegaly is absent (important – if spleen is enlarged, think of another diagnosis).
Diagnosis (diagnosis of exclusion)
- Isolated thrombocytopenia (often <30,000/mm³); Hb and WBC normal.
- Peripheral smear: large platelets (young platelets released to compensate); no schistocytes.
- Normal PT, PTT, fibrinogen.
- Bone marrow (not routinely needed): increased megakaryocytes.
Management
- Platelets >30,000/mm³ & no bleeding → observe (especially children).
- Symptomatic / platelets <30,000 → corticosteroids (prednisone) first line.
- Severe bleeding or need rapid rise → IVIG or anti-D immunoglobulin (Rh+ only).
- Refractory / chronic adult ITP → splenectomy, rituximab, or TPO-receptor agonists (romiplostim, eltrombopag).
- Platelet transfusion only for life-threatening bleeding (transfused platelets are destroyed quickly).
Thrombotic Thrombocytopenic Purpura (TTP)
Definition: Life-threatening thrombotic microangiopathy caused by deficiency of ADAMTS13, the enzyme that cleaves ultra-large von Willebrand factor (vWF) multimers.
Pathophysiology
- Normally ADAMTS13 cuts large vWF multimers into smaller, functional pieces.
- In TTP: ADAMTS13 deficient (autoantibody in adults – acquired; or genetic – Upshaw-Schulman syndrome) → uncleaved ultra-large vWF multimers accumulate.
- These multimers bind platelets → platelet-rich microthrombi in small vessels → consumption of platelets + shearing of RBCs as they pass through (MAHA).
| Mnemonic – TTP Pentad: "FAT RN" | |
Only ~40% of patients have all five – thrombocytopenia + MAHA alone (without another cause) is enough to start treatment. |
جملة تذكرية |
Clinical features
- Adult woman, often 30–50 years old. Triggers: pregnancy, HIV, drugs (clopidogrel, quinine, cyclosporine).
- Neurologic symptoms predominate (vs HUS, which is renal): confusion, headache, focal deficits, seizures.
- Fever, jaundice, fatigue.
Diagnosis
- Severe thrombocytopenia + MAHA: schistocytes on smear, ↑ LDH, ↑ indirect bilirubin, ↓ haptoglobin, ↑ reticulocytes, negative Coombs.
- Normal PT, PTT, fibrinogen → distinguishes from DIC.
- Confirmatory: ADAMTS13 activity <10%.
Management
- Urgent plasma exchange (plasmapheresis) – replaces ADAMTS13 and removes autoantibodies. Start immediately on clinical suspicion.
- Add corticosteroids ± rituximab for acquired TTP.
- Caplacizumab (anti-vWF) in refractory cases.
- Avoid platelet transfusion – adds fuel to the fire (more microthrombi). Only used for life-threatening bleeding.
Hemolytic Uremic Syndrome (HUS)
Definition: Thrombotic microangiopathy presenting with the classic triad: microangiopathic hemolytic anemia + thrombocytopenia + acute kidney injury.
Etiology
- Typical (90%) – Shiga toxin-producing E. coli O157:H7 (STEC-HUS), especially after eating undercooked ground beef or unpasteurized milk. Shigella dysenteriae is another cause.
- Atypical – complement dysregulation (factor H mutation). No diarrhea.
Pathophysiology
- Shiga toxin binds Gb3 receptors on renal endothelium → endothelial injury → platelet activation and microthrombi → mechanical hemolysis (schistocytes) + AKI.
- ADAMTS13 is normal in HUS (unlike TTP).
Clinical features
- Typically a child <5 years with bloody diarrhea 5–10 days earlier.
- Pallor, oliguria, hematuria, edema, hypertension.
- Neurologic symptoms are less prominent than TTP (renal > neuro).
Diagnosis
- CBC: anemia + thrombocytopenia; schistocytes on smear.
- Hemolysis labs: ↑ LDH, ↑ indirect bilirubin, ↓ haptoglobin, negative Coombs.
- ↑ BUN, ↑ creatinine; urinalysis: hematuria, proteinuria.
- Stool culture / Shiga toxin PCR positive.
- Normal PT, PTT, fibrinogen.
Management
- Supportive care is the cornerstone: IV fluids, electrolyte correction, dialysis if needed, RBC transfusion for severe anemia.
- Avoid antibiotics in STEC-HUS – they may increase toxin release and worsen outcome.
- Avoid anti-motility agents (loperamide).
- Atypical HUS → eculizumab (anti-C5 monoclonal antibody).
- Plasma exchange is not first line for typical HUS (unlike TTP).
Heparin-Induced Thrombocytopenia (HIT)
Definition: Immune-mediated, drug-induced thrombocytopenia caused by IgG antibodies against the heparin–platelet factor 4 (PF4) complex. The platelets are activated, not just destroyed, which is why HIT causes thrombosis rather than bleeding.
Two types
- Type I – non-immune, mild (platelets >100,000), within 1–2 days, resolves spontaneously. No clinical significance.
- Type II – immune-mediated, the dangerous form. This is what "HIT" usually refers to.
Pathophysiology (Type II)
- Heparin binds platelet factor 4 (PF4), forming heparin–PF4 complex.
- IgG antibodies form against this complex.
- Immune complex binds platelet FcγRIIa receptors → massive platelet activation and aggregation.
- Result: platelet consumption (thrombocytopenia) + thrombin generation (paradoxical thrombosis).
Clinical features
- Platelet drop >50% from baseline (often nadir 30,000–70,000) 5–10 days after starting heparin.
- Rapid drop within 24 hours possible if patient had heparin exposure in last 100 days.
- Thrombosis (in ~50%): DVT, PE, arterial limb ischemia, stroke, MI, skin necrosis at injection sites.
- More common with unfractionated heparin than with LMWH (enoxaparin).
Diagnosis
- Use the 4T score first (pretest probability):
- Thrombocytopenia magnitude
- Timing of platelet fall
- Thrombosis or other sequelae
- OTher causes excluded
- Score 4–8 → test for anti-PF4 antibodies (ELISA, sensitive screen).
- Serotonin release assay (SRA) – gold standard confirmatory test.
Management
- Stop ALL heparin immediately (including flushes and LMWH).
- Start a non-heparin anticoagulant:
- Direct thrombin inhibitors: argatroban (preferred in renal failure), bivalirudin.
- Fondaparinux or DOACs as alternatives.
- Do NOT give warfarin alone early (causes skin necrosis and worsens thrombosis); bridge with a parenteral non-heparin agent first until platelets >150,000.
- Do NOT transfuse platelets (adds fuel to thrombosis).
Side-by-Side Comparison
Use this table as your single most important review for thrombocytopenia questions. Most exam stems can be solved by matching the patient profile + smear finding to one column.
| ITP vs TTP vs HUS vs HIT – Quick Comparison | ||||
|---|---|---|---|---|
| Feature | ITP | TTP | HUS | HIT |
| Typical patient | Child post-viral / adult woman | Adult woman, 30–50 y | Child <5 y after bloody diarrhea | Patient on heparin 5–10 days |
| Mechanism | Anti-platelet IgG (GpIIb/IIIa) | ADAMTS13 deficiency → large vWF | Shiga toxin (E. coli O157:H7) | IgG vs heparin–PF4 complex |
| Main clinical clue | Isolated bleeding, well-appearing | Neurologic + fever (pentad) | Bloody diarrhea + AKI | New thrombosis on heparin |
| Schistocytes | No | Yes | Yes | No |
| Renal failure | No | Mild | Severe (hallmark) | No |
| Neurologic signs | No | Prominent | Mild/absent | Stroke if thrombosis |
| PT / PTT | Normal | Normal | Normal | Normal |
| Treatment | Steroids, IVIG, splenectomy | Plasma exchange + steroids | Supportive; eculizumab (atypical) | Stop heparin; argatroban / fondaparinux |
| Platelet transfusion | Only if life-threatening bleed | Avoid | Avoid (unless bleeding) | Avoid |
You may also reference the library tables for HUS vs TTP vs ITP differential and the 4T score for HIT.
Mnemonics
| Mnemonics – جمل تذكرية | |
|
جملة تذكرية |
Key Points for Exams
| Key Points for Exams – نقاط مهمة للامتحانات | |
|
تذكر |
احصل على التجربة الكاملة
اشترك للوصول لفيديوهات الشرح التفصيلي والبطاقات التعليمية التفاعلية وأسئلة الممارسة مع تتبع التقدم.