Summary
Hereditary spherocytosis (HS) is the most common inherited hemolytic anemia in people of Northern European descent. It is caused by a defect in red blood cell (RBC) membrane proteins (most often ankyrin or spectrin), which makes RBCs lose their normal biconcave shape and become small, round spherocytes. These abnormal cells are trapped and destroyed in the spleen (extravascular hemolysis).
The classic patient is a child or young adult with the triad of anemia, jaundice, and splenomegaly. Labs show ↑ MCHC, ↑ reticulocytes, ↑ indirect bilirubin, and a negative Coombs test. Diagnosis is confirmed by EMA binding test or osmotic fragility test. Treatment is supportive with folic acid, and splenectomy is curative for severe cases. Main complications are pigmented gallstones and aplastic crisis after parvovirus B19 infection.
Pathophysiology
The core problem in HS is a defect in RBC membrane–cytoskeleton anchoring proteins. Normal RBCs are biconcave because the lipid bilayer is tightly tethered to an internal cytoskeleton by spectrin, ankyrin, band 3, and protein 4.2.
When any of these is defective:
- The bilayer loses anchoring and small membrane vesicles bud off.
- The RBC loses surface area while keeping its volume → it becomes a sphere.
- Spherocytes are rigid and cannot squeeze through splenic sinusoids.
- Macrophages in the splenic red pulp trap and destroy them → extravascular hemolysis.
- Heme is broken down → ↑ unconjugated (indirect) bilirubin → jaundice and pigmented gallstones.
- Bone marrow compensates → ↑ reticulocytes (polychromasia on smear).
Key point: because hemolysis is extravascular, you do not see hemoglobinuria or hemosiderinuria (these are intravascular features).
Polychromasia on smearEpidemiology and Genetics
- Most common inherited hemolytic anemia in people of Northern European ancestry (~1 in 2,000–5,000).
- Inheritance: Autosomal dominant in ~75% of cases (positive family history of anemia, jaundice, or splenectomy). Autosomal recessive and de novo mutations account for the rest.
- Most common mutated gene: ANK1 (ankyrin) — ~50% of cases.
- Other genes: SPTA1, SPTB (spectrin), SLC4A1 (band 3), EPB42 (protein 4.2).
- Age of presentation: wide range — from neonatal jaundice to adulthood, depending on severity.
Clinical Features
The classic triad:
- Anemia — fatigue, pallor, exertional dyspnea.
- Jaundice — scleral icterus, dark urine (urobilinogen, not hemoglobin).
- Splenomegaly — left-upper-quadrant fullness; the spleen is the site of hemolysis.
Other presentations to know:
- Neonatal jaundice — severe, prolonged, may need phototherapy or exchange transfusion. May be the first clue.
- Pigmented (black) gallstones in adolescents/young adults → biliary colic, cholecystitis.
- Aplastic crisis — sudden drop in hemoglobin with low reticulocytes after parvovirus B19 infection.
- Hemolytic crisis — worsening anemia triggered by infection (more common than aplastic crisis, with high reticulocytes).
- Family history of anemia, jaundice, gallstones at a young age, or prior splenectomy.
Diagnosis
Diagnosis combines CBC + smear + hemolysis labs + a confirmatory test.
Initial labs
- CBC: normocytic anemia, ↑ MCHC (> 36 g/dL) — the most specific clue. ↓ MCV is uncommon; MCV is often normal.
- RDW: elevated.
- Reticulocyte count: increased (compensatory).
- Hemolysis panel: ↑ indirect bilirubin, ↑ LDH, ↓ haptoglobin.
- Direct Coombs (DAT): NEGATIVE — this is the key step to rule out autoimmune hemolysis.
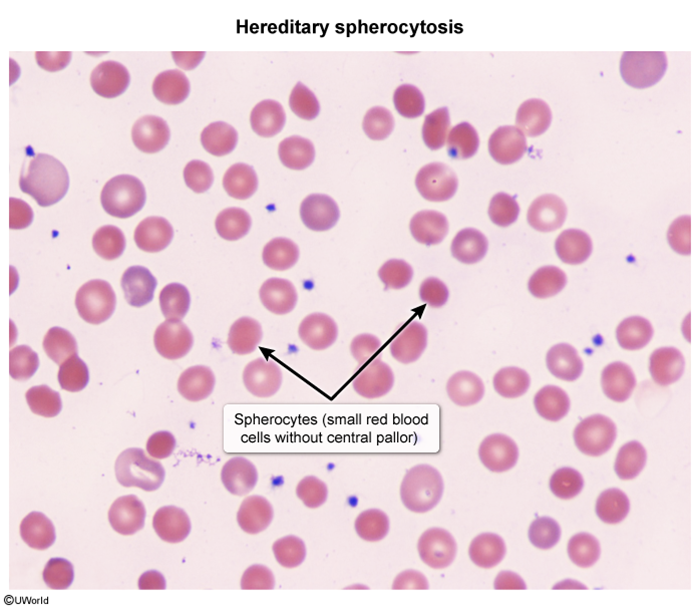
Peripheral smear
Spherocytes: small, round, densely staining RBCs lacking central pallor. Polychromasia (reticulocytes) is also seen.
Confirmatory tests
- EMA (eosin-5-maleimide) binding test — current first-line confirmatory test. ↓ fluorescence because EMA binds band 3 (deficient in HS). Highly sensitive and specific.
- Osmotic fragility test — older classic test. Spherocytes lyse easily in hypotonic saline. Less sensitive than EMA; can miss mild cases.
- Acidified glycerol lysis test — alternative.
Differential Diagnosis
Both HS and warm autoimmune hemolytic anemia (AIHA) show spherocytes — Coombs test separates them.
| Spherocytes on Smear — HS vs. Autoimmune Hemolytic Anemia (AIHA) | ||
|---|---|---|
| Feature | Hereditary Spherocytosis | Warm AIHA |
| Mechanism | Membrane protein defect (genetic) | Autoantibody (IgG) against RBC |
| Coombs (DAT) | NEGATIVE | POSITIVE |
| Family history | Often present | Usually absent |
| Age | Childhood / young adult | Any age; often adult |
| MCHC | ↑ (classic clue) | Usually normal |
| Treatment | Folic acid ± splenectomy | Steroids ± rituximab |
Other conditions to consider:
- G6PD deficiency — X-linked, episodic oxidative hemolysis with Heinz bodies and bite cells after triggers (fava beans, primaquine, sulfa, infection). No spherocytes.
- ABO hemolytic disease of the newborn — neonatal jaundice with spherocytes, but Coombs positive.
- Sickle cell disease / thalassemia — different smear (sickle cells, target cells); abnormal hemoglobin electrophoresis.
- Hereditary elliptocytosis — same family of membrane defects but smear shows elliptocytes, usually mild.
Management
Supportive care (all patients)
- Folic acid supplementation — to support the high RBC turnover and prevent megaloblastic crisis.
- RBC transfusions — for severe anemia, hemolytic or aplastic crises, and symptomatic infants.
- Phototherapy ± exchange transfusion — for severe neonatal jaundice.
- Vaccination before any splenectomy: pneumococcal, meningococcal, H. influenzae type b (encapsulated organisms).
Splenectomy (definitive)
Removes the site of hemolysis → anemia and jaundice resolve, but the membrane defect persists and spherocytes remain on smear.
- Indications: moderate-to-severe anemia, transfusion dependence, growth failure, severe symptoms.
- Timing: ideally after age 5–6 years to reduce risk of overwhelming post-splenectomy infection (OPSI).
- Cholecystectomy is often done at the same time if gallstones are present.
- Post-splenectomy: lifelong penicillin prophylaxis is given to children; watch for sepsis from encapsulated bacteria.
Complications
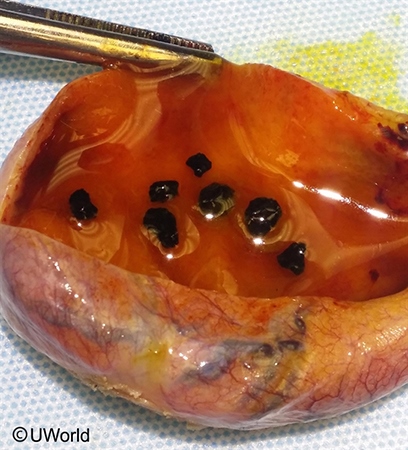
- Pigmented (black) gallstones — chronic hemolysis → ↑ unconjugated bilirubin → calcium bilirubinate stones. May cause biliary colic, cholecystitis, or pancreatitis. Common even in adolescents.
- Aplastic crisis — caused by parvovirus B19. The virus infects erythroid progenitors → sudden marrow shutdown → severe anemia with low reticulocytes. Self-limited but may need transfusion.
- Hemolytic crisis — triggered by infection; ↑ reticulocytes, worsening anemia and jaundice.
- Megaloblastic crisis — folate depletion in patients not on supplements.
- Post-splenectomy sepsis (OPSI) — encapsulated organisms (S. pneumoniae, N. meningitidis, H. influenzae). Prevented by vaccination + prophylactic penicillin in children.
- Iron overload — in transfusion-dependent patients.
Mnemonics
| Mnemonic – HS Key Features: "SPHERE" | |
|
جملة تذكرية |
| Mnemonic – Membrane proteins: "A SPECtacular Band of 4.2" | |
|
جملة تذكرية |
Key Points for Exams
| Key Points for Exams – نقاط مهمة للامتحانات | |
|
تذكر |
احصل على التجربة الكاملة
اشترك للوصول لفيديوهات الشرح التفصيلي والبطاقات التعليمية التفاعلية وأسئلة الممارسة مع تتبع التقدم.