Summary
Thalassemia is an autosomal recessive microcytic, hypochromic anemia (MCV < 80 fL, often < 70 fL) caused by quantitative deficiency of structurally normal globin chains due to gene deletions (alpha-thalassemia, chromosome 16) or point mutations (beta-thalassemia, chromosome 11). Imbalanced chain synthesis causes globin precipitation, leading to ineffective erythropoiesis, peripheral hemolysis, and compensatory marrow expansion (e.g., chipmunk facies, crew-cut skull). Peripheral smear classically reveals target cells, teardrop cells, and basophilic stippling with a normal RDW and normal/high RBC count (Mentzer index < 13).
Diagnosis is confirmed via Hb electrophoresis (beta-thalassemia minor: HbA2 > 3.5%; major: elevated HbF) or genetic testing, alongside normal/high ferritin. Treatment ranges from genetic counseling for traits to chronic transfusions paired with iron chelation (e.g., deferasirox) for major forms; empirical iron supplementation is strictly contraindicated.
Overview
Thalassemia is an inherited (autosomal recessive) microcytic, hypochromic anemia caused by reduced production of a structurally normal globin chain — α (α-thalassemia) or β (β-thalassemia). It is among the most common genetic disorders worldwide, with high prevalence in Mediterranean, Middle Eastern (including Jordan), African, and Southeast Asian populations.
Core genetics (α vs β)
- α-thalassemia → gene DELETION · chromosome 16 · 4 α-genes total (2 per chromosome).
- β-thalassemia → POINT mutation (splicing/promoter defect) · chromosome 11 · 2 β-genes total.
- Quantitative defect: globin chains are normal in structure but reduced in amount → severity ∝ number of genes affected. Normal adult HbA = α₂β₂.
Pathophysiology
Pathophysiology — chain imbalance has two consequences
- Unpaired chains precipitate → membrane damage → ineffective erythropoiesis (precursors die in the marrow) + peripheral hemolysis:
- β-thal: ↓ β → excess α-chains precipitate in marrow precursors.
- α-thal: ↓ α → excess β₄ = HbH (adult) or γ₄ = Hb Barts (fetal) tetramers.
- Chronic anemia → EPO drive → massive marrow expansion (skeletal deformity) + extramedullary hematopoiesis (hepatosplenomegaly).
- Iron overload from ↑ intestinal absorption (hepcidin suppression) + transfusions → the leading cause of death in β-thal major.
The morphologic footprint of ineffective erythropoiesis is visible on the peripheral smear: 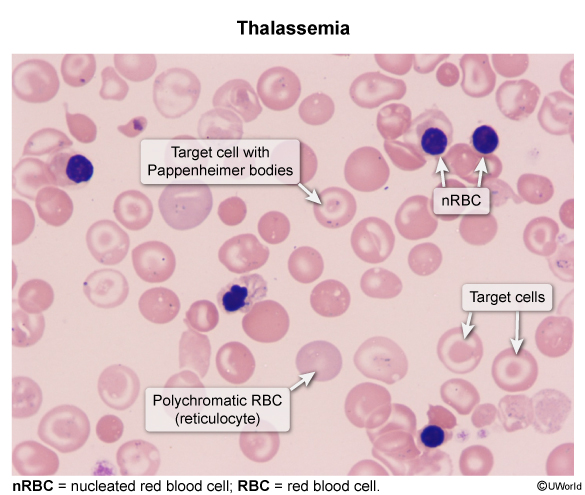
| Mnemonic – جملة تذكرية | |
|
α-thal = “4 A’s”: Asian · African · Absent gene (deletion) · chromosome 16 (= 4 genes). β-thal = “B Med Point”: chromosome 11 (B = 2 genes) · Mediterranean · Point mutation. HbH = Heavy hemolysis (3 α-gene deletion) → Heinz bodies (β₄ precipitates). |
جملة تذكرية |
Clinical Spectrum: α vs. β Thalassemia
Severity follows a gene-dosage spectrum — from a silent carrier to lethal hydrops fetalis. The number of affected globin genes determines where a patient sits on that spectrum.
α-Thalassemia — by number of deleted genes (1–4)
| Alpha-Thalassemia: Genotype, Disorder & Severity: Severity ↑ with each of the 4 α-genes lost (chromosome 16) | |
| 1 gene deleted | Silent carrier (α-thal minima) |
| αα / α– | Asymptomatic; normal CBC; detected only by genetic testing |
| 2 genes deleted | α-thalassemia trait (minor) |
| cis αα/–– (Asian) | Mild microcytic anemia; HIGH risk of hydrops fetalis in offspring |
| trans α–/α– (African) | Mild microcytic anemia; LOW risk for offspring |
| 3 genes deleted | Hemoglobin H disease |
| –– / α– | Moderate–severe hemolytic anemia + splenomegaly; β₄ tetramers (HbH) form Heinz bodies |
| 4 genes deleted | Hb Barts — Hydrops fetalis |
| –– / –– | Incompatible with life; γ₄ (Hb Barts) very high O₂ affinity → tissue hypoxia, anasarca, high-output cardiac failure, death in utero |
- cis deletion = both α-genes lost from the same chromosome (αα/––) → Asian → dangerous for offspring (Hb Barts risk).
- trans deletion = one α-gene from each chromosome (α–/α–) → African → safer.
- Electrophoresis is NORMAL in α-thal trait (the α-chain is shared by HbA, HbA2 and HbF, so all fall proportionately) → diagnosis is by exclusion / genetic testing.
The 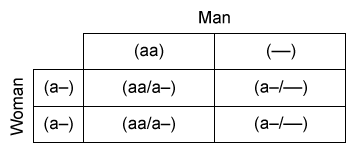 Punnett square illustrates why the spatial arrangement of two deletions drives offspring risk.
Punnett square illustrates why the spatial arrangement of two deletions drives offspring risk.
| فخ امتحاني – Cis vs Trans | |
|
في ثلاسيميا ألفا الصغرى (حذف جينين)، التوضّع cis (αα/––) شائع عند الآسيويين وهو الأخطر على النسل، بينما trans (α–/α–) شائع عند الأفارقة وأقل خطورة. إذا كان أحد الوالدين cis والآخر trans، يرتفع احتمال إنجاب طفل بمرض الهيموغلوبين H، ولا يوجد أي احتمال لإنجاب طفل بثلاسيميا ألفا صغرى. |
ملاحظة |
β-Thalassemia — minor → intermedia → major
| Beta-Thalassemia: Clinical Forms (chromosome 11, point mutations) | |||
|---|---|---|---|
| Feature | Minor (trait) | Intermedia | Major (Cooley anemia) |
| Genotype | β/β⁺ (one defective gene) | β⁺/β⁺ or mild β⁰ | β⁰/β⁰ (both absent) |
| Severity | Asymptomatic / mild microcytic anemia | Moderate anemia; transfusion sometimes needed | Severe; transfusion-dependent |
| Onset | Adult — incidental finding | Childhood | 6–12 months (after HbF ↓) |
| Electrophoresis | ↑ HbA2 (>3.5%), slight ↑ HbF | ↑↑ HbF, ↑ HbA2 | ↑↑↑ HbF, ↑ HbA2, almost no HbA |
| Skeleton | None | Mild | Chipmunk facies, crew-cut skull |
| Treatment | None / genetic counseling | Folate ± occasional transfusion ± chelation | Lifelong transfusion + chelation; HSCT curative |
β-thal symptoms appear after ~6 months of age: fetal HbF (α₂γ₂) needs no β-chains, so the defect is unmasked only as the 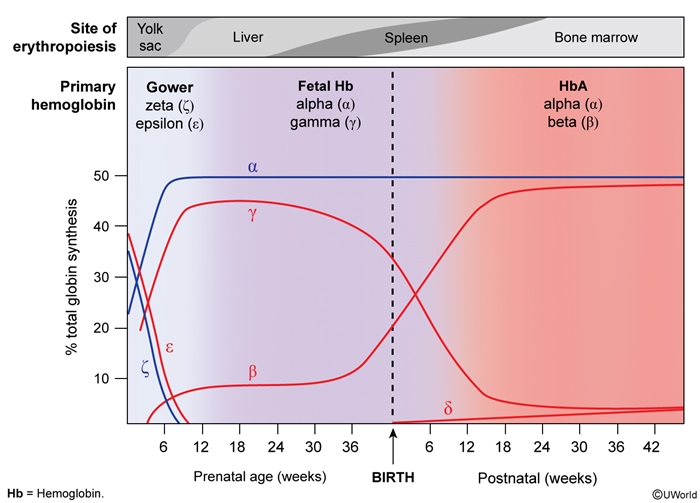 HbF → HbA switch completes. Two alleles set severity: β⁺ (reduced) vs β⁰ (absent) β-chain production.
HbF → HbA switch completes. Two alleles set severity: β⁺ (reduced) vs β⁰ (absent) β-chain production.
See the Alpha Thalassemia genotype–disorder correlation for how each deletion count maps to clinical disease, and the Hemoglobin Electrophoresis patterns in beta-thalassemia for the HbA / HbA2 / HbF percentages that separate minor from major.
Clinical Presentation & Complications
Presentation is a continuum driven by three forces: anemia, chronic hemolysis, and the body's compensatory response (marrow expansion + iron loading). Trait disease is silent; major disease shows the full picture.
Presentation
- Anemia: pallor, fatigue, poor exercise tolerance, tachycardia.
- Chronic hemolysis: jaundice, scleral icterus, dark urine, pigment gallstones.
- Marrow expansion (severe forms): frontal bossing + maxillary overgrowth = "chipmunk facies"; "crew-cut" / "hair-on-end" skull X-ray; pathologic fractures, growth retardation.
- Extramedullary hematopoiesis → hepatosplenomegaly.
Extramedullary hematopoiesis on histology — hematopoietic precursors maturing outside the marrow: 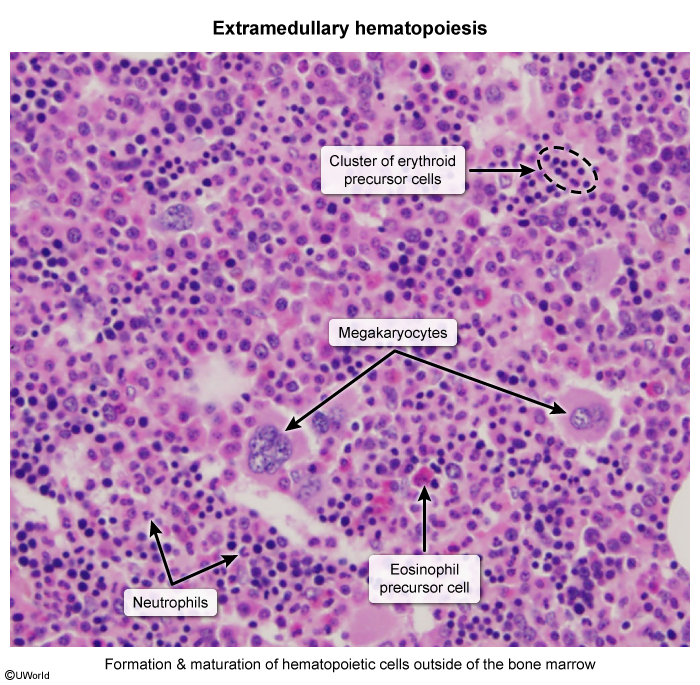
Complications — the severe end of the spectrum
Iron overload (hemosiderosis) from transfusions + ↑ intestinal absorption is the dominant cause of morbidity and mortality:
- Cardiac: dilated cardiomyopathy, arrhythmias, heart failure — most common cause of death.
- Endocrine: diabetes mellitus, hypogonadism (delayed puberty), hypothyroidism, hypoparathyroidism.
- Hepatic / skin: cirrhosis → hepatocellular carcinoma; bronze-grey pigmentation.
Prussian blue (Perl's) staining demonstrates the parenchymal iron behind these complications: 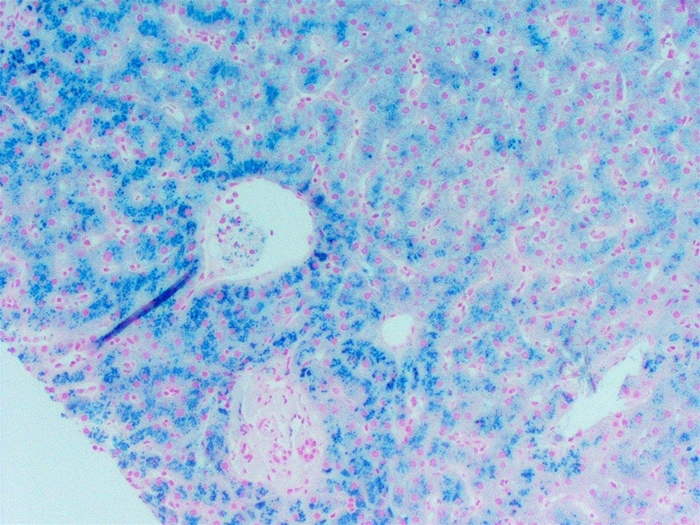
Other complications:
- Hypersplenism → worsening anemia, leukopenia, thrombocytopenia; ↑ thrombosis risk (especially post-splenectomy).
- Hematologic: folate deficiency; aplastic crisis (parvovirus B19).
- Transfusion-related: alloimmunization, infection (HBV/HCV/HIV — now low risk), reactions; paravertebral extramedullary masses can compress the spinal cord (rare, high-yield).
| Important – فكرة سؤال | |
A 6–9 month-old infant of Mediterranean or Middle Eastern descent presents with pallor, failure to thrive, jaundice, hepatosplenomegaly, and frontal bossing. CBC shows severe microcytic anemia; smear shows target cells and nucleated RBCs. ✅ Think → β-Thalassemia Major. Confirm with Hb electrophoresis (↑↑↑ HbF, ↑ HbA2, almost no HbA). Onset at 6–12 months is the clue — it coincides with the physiologic HbF → HbA switch. |
تذكر |
| Note – ملاحظة | |
Iron overload is the main cause of death in β-thalassemia major. Cardiac siderosis → dilated cardiomyopathy and arrhythmias is the leading cause of mortality. Chelation therapy plus cardiac MRI T2* monitoring dramatically improve survival — the iron comes from both chronic transfusions and increased intestinal absorption (hepcidin suppression). |
ملاحظة |
Diagnostic Approach
Any patient with a low MCV enters the microcytic-anemia differential. Workup is stepwise, and two numbers — the RBC count/RDW and the HbA2 level — do most of the discriminating.
Stepwise workup
- Step 1 — CBC: ↓ Hb; ↓↓ MCV (often < 70 fL); normal or HIGH RBC count; normal RDW (cells are uniformly small).
- Step 2 — Peripheral smear:
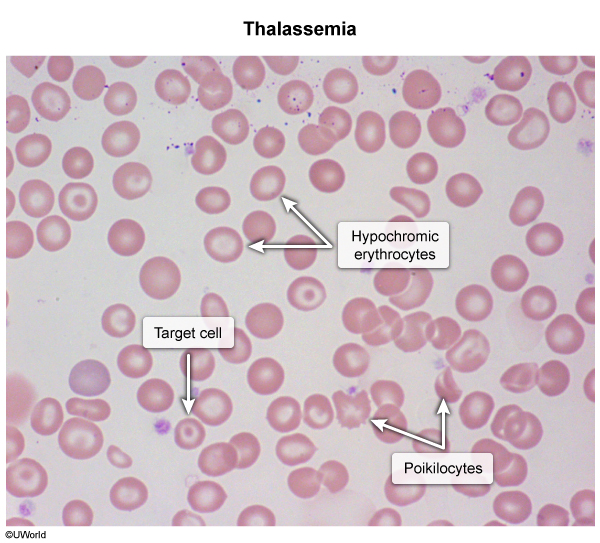 target cells, microcytic-hypochromic RBCs, basophilic stippling, teardrop & nucleated RBCs.
target cells, microcytic-hypochromic RBCs, basophilic stippling, teardrop & nucleated RBCs. - Step 3 — Iron studies: normal or elevated iron, ferritin, transferrin saturation → excludes IDA.
- Step 4 — Hb electrophoresis (HPLC): β-thal minor ↑ HbA2 (>3.5%); β-thal major ↑↑↑ HbF; α-thal trait normal; HbH disease shows a fast-migrating HbH (β₄) band.
- Step 5 — Genetic testing: confirms α-thal and enables prenatal diagnosis / counseling.
Two critical discriminators
- Mentzer index = MCV ÷ RBC count: < 13 = thalassemia; > 13 = iron deficiency.
- HbA2 > 3.5% = β-thalassemia minor (the single most testable lab).
- Never give iron empirically to a microcytic patient who may have thalassemia — risk of iron overload.
| Master Differential of Microcytic Anemia | |||
|---|---|---|---|
| Parameter | Thalassemia Trait | Iron Deficiency (IDA) | Anemia of Chronic Disease |
| MCV | ↓↓ (very low) | ↓ | ↓ or normal |
| RDW | Normal | ↑ (high) | Normal |
| RBC count | Normal / ↑ | ↓ | ↓ |
| Serum iron | Normal | ↓ | ↓ |
| Ferritin | Normal / ↑ | ↓↓ | ↑ (high) |
| TIBC | Normal | ↑ | ↓ |
| Hb electrophoresis | ↑ HbA2/HbF (β-thal); normal (α-thal) | Normal | Normal |
| Best clue | Mentzer index < 13; HbA2 > 3.5% | Mentzer > 13; ↓ ferritin | Underlying chronic illness |
Refer to the iron studies in microcytic anemia for the ferritin / TIBC / transferrin-saturation pattern that keeps thalassemia (normal/high iron) apart from IDA, and the Hemoglobin Electrophoresis patterns in beta-thalassemia for the diagnostic HbA2 / HbF percentages.
| فخ امتحاني – Thalassemia vs IDA | |
|
أهم ما يميّز الثلاسيميا عن فقر الدم بعوز الحديد هو أن تعداد الكريات الحمر (RBC count) يكون طبيعياً أو مرتفعاً في الثلاسيميا مع RDW طبيعي، بينما يكون منخفضاً مع ارتفاع الـ RDW في عوز الحديد. مؤشر Mentzer < 13 يرجّح الثلاسيميا. ولا تعطِ الحديد لمريض الثلاسيميا إلا بعد إثبات العوز مخبرياً. |
ملاحظة |
Management & Prevention
Treatment is matched to disease severity. Trait needs counseling only; intermedia needs supportive care; major needs the transfusion–chelation backbone, with HSCT as the only cure.
Minor / trait
- No treatment needed.
- Genetic counseling + premarital screening = the key step (especially in consanguineous populations).
- Do NOT give iron unless deficiency is lab-proven.
Intermedia
- Folic acid supplementation daily.
- Occasional transfusion for symptomatic anemia or growth failure.
- Monitor ferritin; chelate if iron overload develops.
Major (and severe HbH disease)
- Step 1 — Chronic transfusion: maintain Hb > 9–10 g/dL to suppress erythropoiesis and prevent skeletal deformity.
- Step 2 — Iron chelation: start once ferritin > 1000 ng/mL or after ~10–20 transfusions (agents below).
- Step 3 — Folic acid daily for the increased demand of chronic hemolysis.
- Step 4 — Splenectomy: if transfusion requirement rises sharply or hypersplenism develops; vaccinate against encapsulated organisms first (S. pneumoniae, H. influenzae, N. meningitidis).
- Step 5 — Allogeneic HSCT: the only curative therapy; best results in young patients with an HLA-matched sibling donor.
- Step 6 — Newer options: luspatercept (improves erythroid maturation, ↓ transfusion need in adults); gene therapy emerging for transfusion-dependent disease.
| Iron Chelation Agents (transfusion-dependent disease) | |||
|---|---|---|---|
| Agent | Deferasirox | Deferiprone | Deferoxamine |
| Route | Oral (first-line) | Oral | IV / SC infusion |
| Dose | 20–40 mg/kg once daily | 25 mg/kg three times daily | 20–60 mg/kg SC over 8–12 h, 5–7 nights/week |
| Key toxicity | Renal impairment, GI upset, ↑ LFTs | Agranulocytosis (monitor CBC weekly) | Ototoxicity, retinopathy, heavy infusion burden |
See the Red Blood Cell Transfusion Thresholds by Hemoglobin Level for the Hb cutoffs that guide when to transfuse.
| نقطة وقائية – Prevention | |
|
في الأردن والشرق الأوسط، حيث يشيع زواج الأقارب، يُعدّ الفحص ما قبل الزواج (premarital screening) والاستشارة الوراثية أهم تدخل وقائي للصحة العامة لتقليل ولادة أطفال مصابين بالثلاسيميا الكبرى. |
ملاحظة |
احصل على التجربة الكاملة
اشترك للوصول لفيديوهات الشرح التفصيلي والبطاقات التعليمية التفاعلية وأسئلة الممارسة مع تتبع التقدم.