Summary
Primary myelofibrosis (PMF) is a chronic, BCR-ABL-negative myeloproliferative neoplasm driven by constitutive JAK-STAT activation (JAK2 V617F, CALR, or MPL mutations). Clonal megakaryocytes secrete fibrogenic cytokines (TGF-beta), driving reactive reticulin and collagen marrow fibrosis, progressive marrow failure, and extramedullary hematopoiesis. Patients present with constitutional symptoms and massive splenomegaly. Peripheral smear shows a leukoerythroblastic picture with pathognomonic teardrop cells (dacrocytes). Bone marrow aspiration characteristically yields a dry tap, making core bone marrow biopsy with reticulin staining the gold-standard diagnostic test. Management for intermediate/high-risk disease utilizes ruxolitinib (JAK1/2 inhibitor) for splenomegaly and symptom control; allogeneic stem cell transplantation is the sole curative therapy. Feared complications include transformation to acute myeloid leukemia.
Overview
Myelofibrosis (MF) is a chronic, BCR-ABL–negative myeloproliferative neoplasm (MPN) in which a clonal hematopoietic stem cell (predominantly the megakaryocyte line) drives progressive replacement of the bone marrow by fibrous tissue (reticulin and collagen). As the marrow fails, blood production shifts to the spleen and liver (extramedullary hematopoiesis), producing the disease's hallmark — massive splenomegaly.
- Primary MF (PMF): arises de novo.
- Secondary MF: evolves from polycythemia vera (post-PV MF) or essential thrombocythemia (post-ET MF).
Epidemiology
- Age: typically > 60 years (median ~65).
- Sex: slight male predominance.
- Incidence: the rarest of the classical MPNs (~1 per 100,000/year).
- Risk factors: prior radiation or benzene exposure; antecedent PV or ET.
Pathophysiology
Driver mutations
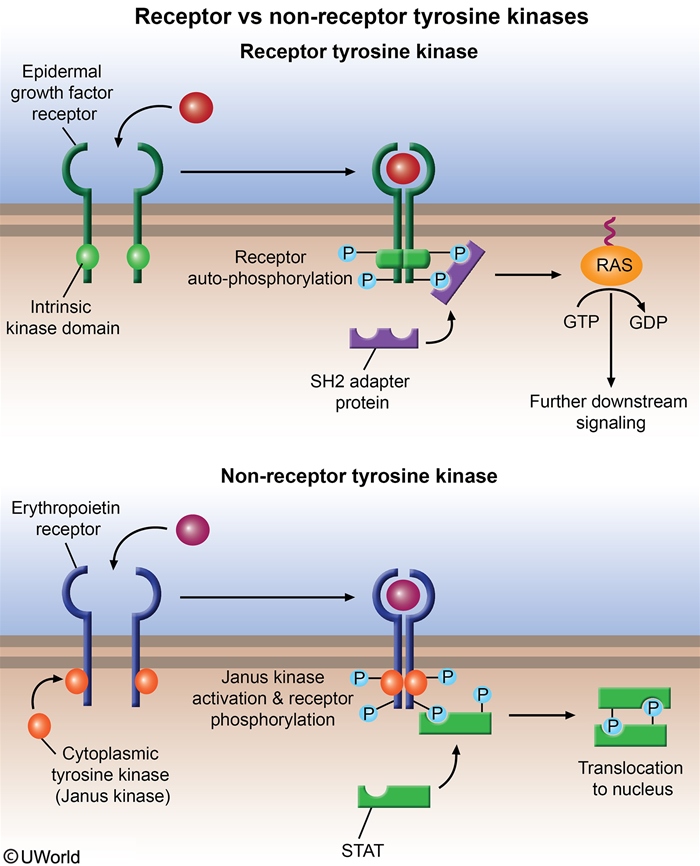
MF is driven by mutations that constitutively activate the JAK-STAT signaling pathway, producing uncontrolled myeloid proliferation independent of cytokine binding.
- JAK2 V617F — most common (~50–60%).
- CALR (calreticulin) — ~25%; best prognosis.
- MPL (thrombopoietin receptor) — ~5–10%.
- "Triple-negative" (~10%) — worst prognosis.
Crucially, MF is BCR-ABL negative — distinguishing it from CML, which is defined by the Philadelphia chromosome t(9;22).
| Mnemonic – MPN driver mutations | |
|
"JAK of all trades" — JAK2 is mutated in all three classical Philadelphia-negative MPNs:
Only CML breaks the pattern → BCR-ABL t(9;22). |
جملة تذكرية |
Core mechanism
The disease starts as a clonal stem cell disorder, but the fibrosis itself is reactive and non-clonal — a secondary response to abnormal megakaryocytes:
- Clonal megakaryocyte proliferation (driven by JAK-STAT activation).
- Megakaryocytes secrete cytokines: TGF-β, PDGF, bFGF, VEGF.
- TGF-β stimulates marrow fibroblasts to deposit reticulin and collagen.
- Fibrotic marrow can no longer support hematopoiesis → progressive cytopenias.
- Hematopoiesis relocates to the spleen and liver → massive splenomegaly + hepatomegaly.
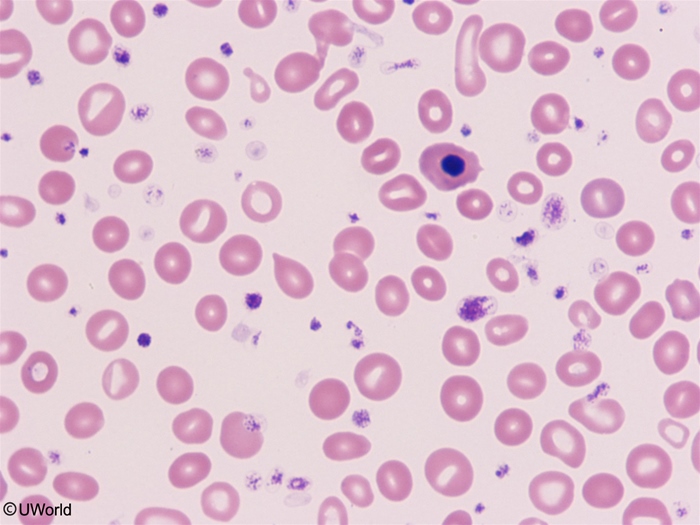
- RBCs are mechanically deformed squeezing through fibrotic marrow and the enlarged spleen → teardrop cells (dacrocytes).
The pathogenesis and clinical manifestations of primary myelofibrosis reference table lays out how clonal megakaryocytes, TGF-β, and extramedullary hematopoiesis connect to the bedside findings.
Clinical Presentation & Course
Onset is usually insidious. Up to 30% of patients are asymptomatic at diagnosis, discovered incidentally with splenomegaly or an abnormal CBC. When symptomatic, the picture is dominated by a classic triad:
1. Constitutional symptoms (high circulating cytokines)
- Fatigue (often profound and activity-limiting), weight loss, low-grade fever, night sweats, pruritus.
2. Massive splenomegaly (extramedullary hematopoiesis) — the hallmark
- Massive splenomegaly, often crossing the midline.
- Early satiety, left-upper-quadrant fullness/pain, and splenic infarcts.
- Hepatomegaly from hepatic extramedullary hematopoiesis.
3. Progressive cytopenias (marrow failure)
- Anemia → pallor, exertional dyspnea (frequently transfusion-dependent later).
- Thrombocytopenia → bleeding in advanced disease.
- WBC count variable — high early, then normal or low.
Other findings: bone pain and gout / hyperuricemia from high cell turnover.
| Important – فكرة سؤال | |
|
An elderly patient presents with fatigue, weight loss, early satiety, and a huge spleen. The blood smear shows teardrop cells and nucleated RBCs (leukoerythroblastic picture), and bone marrow aspirate gives a "dry tap" → think primary myelofibrosis. |
تذكر |
Complications & course
- Transformation to acute myeloid leukemia (AML) — in ~10–20%; carries a very poor prognosis.
- Severe anemia with transfusion dependence.
- Infections (neutropenia) and bleeding (thrombocytopenia / platelet dysfunction).
- Thrombosis — including Budd-Chiari syndrome and portal vein thrombosis.
- Portal hypertension from hepatic extramedullary hematopoiesis and increased splenoportal flow.
- Gout / urate nephropathy from high cell turnover.
MPNs are a recognized cause of hepatic vein thrombosis — see the Budd-Chiari syndrome etiology and workup for why JAK2 testing belongs in the workup of an unexplained hepatic vein thrombosis.
Prognosis: median survival ~5 years overall, but ranges widely — from <2 years (high-risk) to >10 years (low-risk) — and is formally estimated with the DIPSS / DIPSS-Plus scoring systems.
Diagnostic Approach
Diagnosis follows a stepwise workflow: CBC + peripheral smear → bone marrow biopsy → molecular testing, while excluding mimics of marrow fibrosis.
1. CBC & peripheral blood smear
- Anemia (usually normocytic); WBC and platelets variable (often high early, then low).
- Leukoerythroblastic picture = immature WBCs (myelocytes/metamyelocytes) + nucleated RBCs.
- Teardrop cells (dacrocytes) — classic and high-yield.
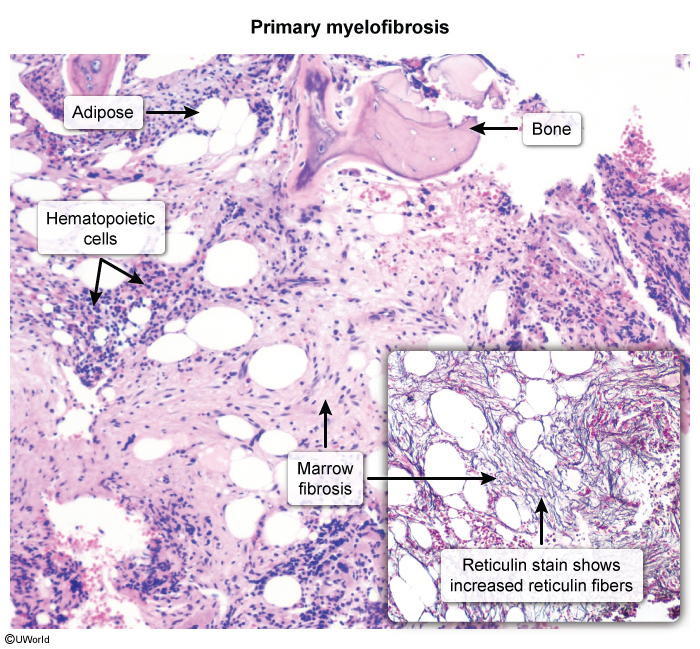
2. Bone marrow
- Aspirate → "dry tap" (aspiration fails because of fibrosis).
- Biopsy is the diagnostic step: hypercellular early, then fibrotic marrow with reticulin/collagen deposition (highlighted on reticulin stain) and clusters of atypical megakaryocytes.
| ملاحظة سريرية – Clinical Note | |
الشفط الجاف (dry tap) مع وجود خلايا دمعية (dacrocytes) على لطاخة الدم يوجه بقوة نحو تليف نقوي العظم، والخطوة التالية هي أخذ خزعة (biopsy) وليس إعادة الشفط. |
ملاحظة |
3. Molecular & laboratory workup
- JAK2, CALR, MPL mutation testing (a driver mutation supports clonality).
- BCR-ABL testing → must be negative to exclude CML.
- Supportive labs: ↑ LDH, ↑ uric acid, ↑ ALP.
Differential diagnosis
The key task is separating PMF from the other MPNs and from secondary marrow fibrosis:
- Other MPNs (CML, PV, ET) — compared below.
- Secondary myelofibrosis: metastatic marrow infiltration, lymphoma, infections (TB), autoimmune disease (SLE), and hairy cell leukemia (also massive splenomegaly + dry tap, but distinguished by flow cytometry).
- Acute leukemia — consider when blasts rise.
- Aplastic anemia — pancytopenia but marrow is hypocellular without fibrosis.
| Chronic Myeloproliferative Neoplasms — Master Comparison | ||||
|---|---|---|---|---|
| Feature | CML | PV | ET | PMF |
| Main cell line | Granulocytes | RBCs | Platelets | Megakaryocytes → fibrosis |
| Driver mutation | BCR-ABL t(9;22) | JAK2 (~95%) | JAK2 (~50%) / CALR / MPL | JAK2 / CALR / MPL |
| Splenomegaly | Moderate–massive | Moderate | Mild | Massive (hallmark) |
| Classic smear | All myeloid stages, basophilia | ↑↑ RBCs, plethora | ↑↑ platelets, giant forms | Teardrop cells, leukoerythroblastic |
| Bone marrow | Hypercellular, all stages | Hypercellular (panmyelosis) | Large/giant megakaryocytes | Fibrosis, "dry tap" |
| First-line therapy | Imatinib (TKI) | Phlebotomy + ASA ± hydroxyurea | ASA ± hydroxyurea | Ruxolitinib; allo-HSCT if eligible |
Management Principles
Management is risk-adapted using the DIPSS / DIPSS-Plus scores. Apart from transplant, all therapy is symptom-directed and non-curative — the goals are to control splenomegaly, relieve constitutional symptoms, and support cytopenias.
Low-risk / asymptomatic
- Observation ("watch and wait") with periodic monitoring.
Intermediate–high risk / symptomatic
- Ruxolitinib — first-line. Oral JAK1/JAK2 inhibitor that reduces splenomegaly and constitutional symptoms (works regardless of JAK2 mutation status).
- Hydroxyurea — for high WBC/platelet counts or symptomatic splenomegaly not requiring a JAK inhibitor.
- Anemia management: transfusions, erythropoiesis-stimulating agents, danazol, or thalidomide/lenalidomide.
- Splenectomy — reserved for refractory painful splenomegaly or transfusion-dependent anemia.
- Splenic irradiation — for symptomatic splenomegaly in poor surgical candidates.
Curative
- Allogeneic hematopoietic stem cell transplant (allo-HSCT) — the only curative option; reserved for younger, fit, intermediate-2/high-risk patients.
| Note – ملاحظة | |
Ruxolitinib controls symptoms but does not cure or reliably reverse fibrosis. Eligible high-risk patients should still be referred for allo-HSCT — the only therapy that alters the natural history of the disease. |
ملاحظة |
High-Yield Summary & Exam Pearls
Rapid-review of the most testable concepts in myelofibrosis:
- Identity: chronic BCR-ABL–negative MPN; clonal megakaryocytes secrete TGF-β → reactive (non-clonal) marrow fibrosis.
- Mutations: JAK2 V617F (most common, ~50–60%) > CALR (~25%, best prognosis) > MPL; triple-negative = worst prognosis.
- Smear: dacrocytes (teardrop cells) + leukoerythroblastic picture (nucleated RBCs + immature WBCs).
- Marrow: "dry tap" on aspirate → diagnosis requires core biopsy with reticulin stain showing fibrosis + atypical megakaryocyte clusters.
- Hallmark exam: massive splenomegaly (extramedullary hematopoiesis) crossing the midline.
- Treatment: ruxolitinib (JAK1/2 inhibitor) first-line for spleen/symptoms; allo-HSCT is the only cure.
- Feared complication: transformation to AML (~10–20%).
| فخ امتحاني – Exam Trap | |
غياب كروموسوم فيلادلفيا (BCR-ABL) ضروري لتشخيص تليف نقوي العظم الأولي واستبعاد ابيضاض الدم النقوي المزمن (CML). |
ملاحظة |
| Mnemonic – Myelofibrosis essentials | |
"FIBROSIS"
|
جملة تذكرية |
| Key Points for Exams – نقاط مهمة للامتحانات | |
|
ملاحظة |
احصل على التجربة الكاملة
اشترك للوصول لفيديوهات الشرح التفصيلي والبطاقات التعليمية التفاعلية وأسئلة الممارسة مع تتبع التقدم.