Summary
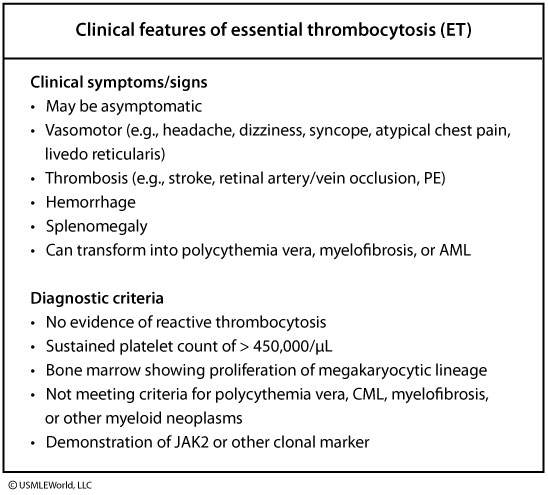
Essential Thrombocythemia (ET) is an indolent, Philadelphia-negative myeloproliferative neoplasm characterized by clonal megakaryocyte proliferation and sustained thrombocytosis (platelets > 450,000/mcL), driven by mutually exclusive JAK2 V617F (~50-60%), CALR (~20-25%), or MPL (~3-5%) mutations. Patients present with arterial/venous thrombosis or vasomotor symptoms like erythromelalgia (burning hand/foot pain relieved by aspirin). Uniquely, extreme thrombocytosis (> 1,000,000/mcL) causes paradoxical mucocutaneous bleeding via acquired von Willebrand disease. Excisional/core bone marrow biopsy demonstrating large, hyperlobulated ("stag-horn") megakaryocytes alongside driver mutation confirmation establishes the diagnosis after excluding reactive causes and CML (BCR-ABL negative). First-line management employs low-dose aspirin for low-risk patients (held if platelets > 1,000,000/mcL) and hydroxyurea cytoreduction for high-risk patients (age > 60 or prior thrombosis).
Overview
Definition
- Essential Thrombocythemia (ET) = a chronic Philadelphia-negative myeloproliferative neoplasm (MPN) defined by sustained thrombocytosis (platelets >450 × 10⁹/L) from clonal megakaryocyte proliferation.
- A JAK-STAT–driven MPN in the same family as Polycythemia Vera and Primary Myelofibrosis — and the most indolent of the three.
Epidemiology
- Incidence: ~1–2 per 100,000/year.
- Age: bimodal — main peak 50–60 yrs, second peak in young women (~30 yrs).
- Sex: slight female predominance.
Top 3 driver mutations (mutually exclusive in ~90%)
| Driver Mutations in Essential Thrombocythemia (mutually exclusive in ~90%) | |||
|---|---|---|---|
| Feature | JAK2 V617F | CALR | MPL |
| Frequency | ~50–60% (most common) | ~20–25% | ~3–5% |
| Mechanism | Constitutive JAK-STAT activation, TPO-independent | Mutant calreticulin activates the MPL receptor → JAK-STAT | Activating thrombopoietin-receptor mutation → JAK-STAT |
| Clinical implication | Older patients; higher thrombosis risk | Younger patients, higher platelet counts, LOWER thrombosis risk, BETTER prognosis | Often older; may be mildly anemic |
- ~10–15% are "triple-negative" (no identifiable driver).
- CALR pearl: CALR-mutated ET = better prognosis and lower thrombosis risk than JAK2.
Pathophysiology
Pathophysiology
- Driver mutation → constitutive JAK-STAT activation → megakaryocytes proliferate independent of thrombopoietin.
- Massive platelet release → hyperviscous, prothrombotic blood.
- Paradox: at very high counts (>1,000–1,500 × 10⁹/L), platelets adsorb large vWF multimers → acquired von Willebrand disease → bleeding.
The shared molecular lesion is constitutive JAK-STAT signalling, illustrated in the JAK2 mutation pathway diagram below.
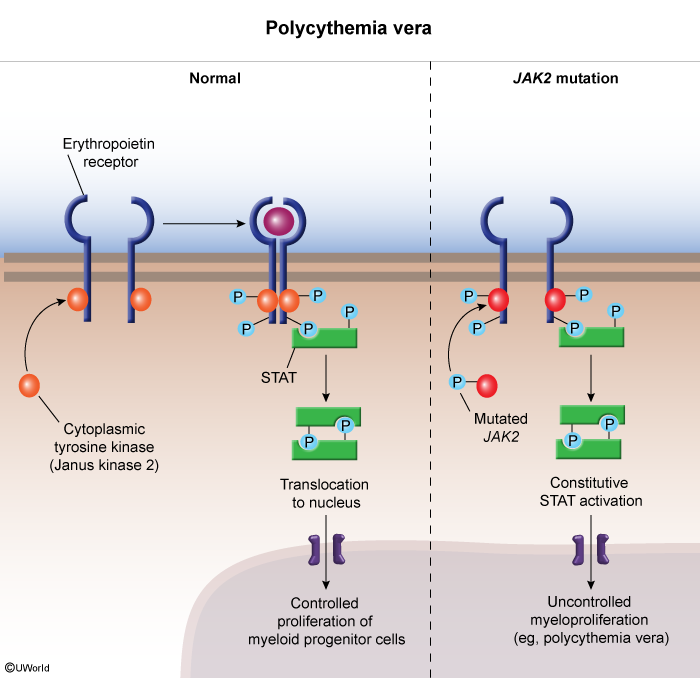
To separate ET from the other clonal myeloid neoplasms, see the four chronic myeloproliferative disorders for the diagnostic features and mutations that distinguish ET from CML, PV, and PMF.
Clinical Presentation
Most patients are asymptomatic at diagnosis (incidental CBC). Symptomatic disease follows a classic triad.
1. Vasomotor / microvascular symptoms
- Erythromelalgia — burning, red, painful hands/feet, dramatically relieved by aspirin (the classic clue).
- Headache, dizziness, transient visual disturbances, paresthesias, tinnitus.
2. Thrombosis (arterial > venous)
- Arterial: stroke / TIA, MI, peripheral arterial occlusion.
- Venous: DVT, PE, and splanchnic vein thrombosis (Budd-Chiari, portal vein).
Splanchnic clots in a young patient are a red flag — refer to the Budd-Chiari syndrome workup for the hepatic-vein outflow obstruction picture and its JAK2 testing.
| ملاحظة سريرية – Clinical Note | |
| أي مريض شاب لديه خثار في الوريد البابي أو الكبدي، يجب فحص طفرة JAK2 لاستبعاد كثرة الصفيحات الأساسية. | ملاحظة |
3. Paradoxical bleeding (platelets >1,000 × 10⁹/L)
- Mucocutaneous bleeding from acquired von Willebrand disease (platelets adsorb large vWF multimers).
- Use aspirin cautiously at very high counts.
| فخ امتحاني – Exam Trap | |
| ارتفاع الصفائح الشديد (>١٠٠٠) يسبب نزيفاً وليس تخثراً بسبب استهلاك عامل فون ويلبراند. | ملاحظة |
Exam findings
- Mild splenomegaly in ~25–50%.
- No significant hepatomegaly and no lymphadenopathy.
Diagnostic Approach
Workup is step-wise: confirm true thrombocytosis → exclude reactive causes → molecular & marrow studies.
Step 1 — Confirm true thrombocytosis
- CBC: platelets persistently >450 × 10⁹/L (≥2 occasions).
- Peripheral smear: increased, often giant/abnormal platelets; WBC and RBC usually normal.
Step 2 — Exclude reactive (secondary) thrombocytosis
- Iron studies (iron deficiency) and CRP/ESR (infection, inflammation).
- History: recent bleeding, surgery, splenectomy, or malignancy.
- Reactive thrombocytosis is the single most common cause of a high platelet count (IL-6 → hepatic thrombopoietin) — exclude it first.
Step 3 — Molecular & bone marrow studies
- Driver panel: JAK2 V617F → CALR → MPL.
- BCR-ABL must be negative (excludes CML).
- BM biopsy: hypercellular with large, mature, hyperlobulated ("stag-horn") megakaryocytes; no significant reticulin fibrosis.
WHO criteria (all 4 major required)
- Platelets >450 × 10⁹/L.
- BM megakaryocyte proliferation with large, mature forms.
- Does not meet criteria for CML, PV, PMF, MDS, or other myeloid neoplasm.
- JAK2, CALR, or MPL mutation (a clonal marker / absence of reactive cause serves as the minor criterion when criterion 4 is absent).
Centerpiece: ET vs. reactive thrombocytosis
| ET vs. Reactive Thrombocytosis | ||
|---|---|---|
| Feature | Essential Thrombocythemia (ET) | Reactive (Secondary) |
| Cause | Clonal JAK2 / CALR / MPL mutation | Infection, inflammation, iron deficiency, post-splenectomy, malignancy |
| Platelet count | Often >600–1,000 × 10⁹/L, persistent | Usually <1,000 × 10⁹/L, transient |
| Platelet morphology | Giant / abnormal forms | Normal |
| Megakaryocytes (BM) | Increased, large, hyperlobulated (stag-horn) | Normal |
| Splenomegaly | May be present (~25–50%) | Absent |
| Thrombosis / erythromelalgia | Yes | No |
| JAK2 / CALR / MPL | Positive in ~90% | Negative |
To separate ET from CML and the other clonal myeloid neoplasms, see the four chronic myeloproliferative disorders for the mutation-based differential.
Management
Treatment is risk-stratified; ET is not curable (except by transplant) but very treatable. Goals: prevent thrombosis, control bleeding, relieve symptoms.
Risk stratification (IPSET, simplified)
- High risk: age >60 OR prior thrombosis (± JAK2-positive with cardiovascular risk factors).
- Low risk: none of the above.
| نقطة قرار علاجي – Treatment Decision | |
| لا نبدأ العلاج الخلوي (Hydroxyurea) إلا للمرضى ذوي الخطورة العالية (عمر >٦٠ أو جلطة سابقة). | ملاحظة |
Treatment by risk
| Risk-Stratified Therapy in ET | ||
|---|---|---|
| Risk group | First-line therapy | Key notes |
| Low risk (age ≤60 AND no prior thrombosis) | Low-dose aspirin 75–100 mg PO daily (± observation) | Aspirin controls vasomotor symptoms (erythromelalgia). HOLD aspirin if platelets >1,000–1,500 × 10⁹/L (bleeding risk from acquired vWD) |
| High risk (age >60 OR prior thrombosis) | Aspirin + cytoreduction | Cytoreduction target: platelets <400 × 10⁹/L |
| Cytoreductive choice | Hydroxyurea 500 mg–1 g PO daily (first-line) | Pegylated interferon-α for young patients / pregnancy; anagrelide is second-line |
Cytoreductive agents (rule of 3)
- Hydroxyurea 500 mg–1 g PO daily — first-line for most high-risk patients.
- Pegylated interferon-α — preferred in pregnancy and young patients (non-teratogenic, non-leukemogenic).
- Anagrelide — second-line; inhibits megakaryocyte maturation (side effects: palpitations, headache, fluid retention).
| Note – ملاحظة | |
| Pregnancy & ET: increased risk of miscarriage and placental thrombosis. Use low-dose aspirin throughout pregnancy; add LMWH postpartum. If cytoreduction is needed → interferon-α (hydroxyurea is teratogenic). | ملاحظة |
Complications & Prognosis
Complications (rule of 3)
- Thrombosis — leading cause of morbidity (stroke, MI, DVT, splanchnic vein thrombosis).
- Bleeding — when platelets >1,000–1,500 × 10⁹/L (acquired von Willebrand disease).
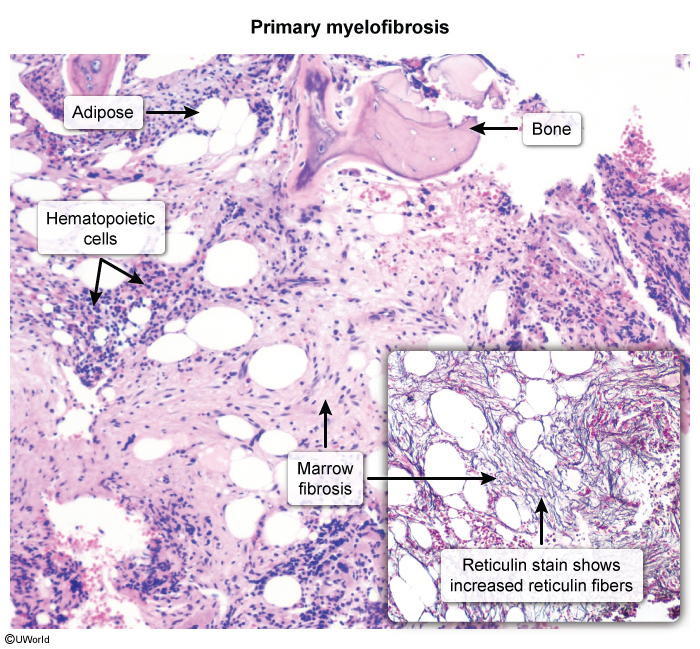
- Transformation (late, uncommon): post-ET myelofibrosis (~5–10% at 15 yrs) or AML (~1–5%, higher with alkylating agents).
Transformation to post-ET myelofibrosis produces the same fibrotic marrow seen in primary myelofibrosis:
Prognosis
- Most indolent MPN; median survival approaches that of the general population if well-managed.
- CALR-mutated patients fare better than JAK2-mutated patients.
- Additional risk: pregnancy loss from placental thrombosis.
احصل على التجربة الكاملة
اشترك للوصول لفيديوهات الشرح التفصيلي والبطاقات التعليمية التفاعلية وأسئلة الممارسة مع تتبع التقدم.