Summary
Polycythemia Vera (PV) is a chronic, BCR-ABL-negative myeloproliferative neoplasm driven by an activating JAK2 V617F mutation that causes erythropoietin (EPO)-independent erythropoiesis. Elevated red blood cell mass leads to hyperviscosity and thrombosis, classically presenting with facial plethora, aquagenic pruritus, splenomegaly, erythromelalgia, and unexplained Budd-Chiari syndrome. Diagnostic hallmarks on initial labs include elevated hemoglobin/hematocrit alongside low serum EPO levels (discriminating primary from secondary polycythemia); gold-standard confirmation requires JAK2 mutation testing and bone marrow biopsy demonstrating trilineage panmyelosis. First-line management for all patients is therapeutic phlebotomy (target hematocrit < 45%) and low-dose aspirin; high-risk patients (age >= 60 or prior thrombosis) require cytoreduction with hydroxyurea. Therapeutic iron deficiency should not be supplemented.
Overview
Polycythemia Vera (PV) is a chronic, BCR-ABL–negative myeloproliferative neoplasm (MPN) in which a clonal hematopoietic stem cell — driven by an activating JAK2 mutation — overproduces red cells (and often platelets and granulocytes) independent of erythropoietin (EPO). The result is a rising RBC mass → hyperviscosity → a strong tendency to thrombosis. The classic patient is an older adult with a ruddy face, itching after a hot shower, splenomegaly, and an unexpectedly high hemoglobin with a low serum EPO.
- Labs at a glance: ↑ Hb/Hct, often ↑ WBC and platelets (panmyelosis), ↓ serum EPO, positive JAK2.
- Treatment at a glance: phlebotomy + low-dose aspirin for everyone; add hydroxyurea if high risk.
- Long-term risks: thrombosis (leading cause of death), progression to myelofibrosis or AML.
Where PV fits
PV belongs to the BCR-ABL–negative MPNs, together with essential thrombocythemia (ET) and primary myelofibrosis (PMF) — all three share constitutive JAK2 signaling. "Polycythemia" simply means too many red cells; the diagnostic task is to separate true clonal PV from secondary (EPO-driven) and relative (volume-contraction) causes. Refer to the chronic myeloproliferative disorders comparison for the mutations and features that separate CML, ET, PV, and PMF.
Epidemiology
- Age: typically > 60 years (median ~60); rare under 40.
- Sex: slight male predominance.
- Incidence: ~1–2 per 100,000 per year; mostly sporadic.
Pathophysiology
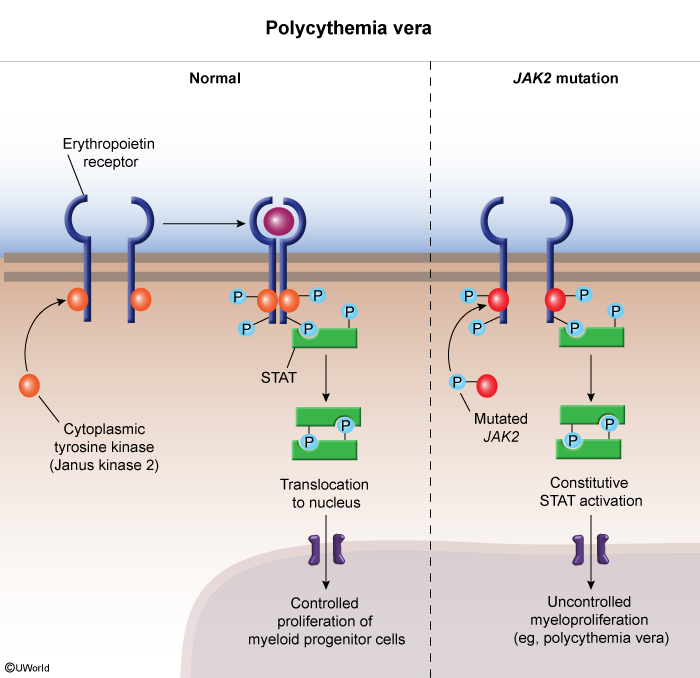
Pathophysiology — JAK2-driven, EPO-independent erythropoiesis
JAK2 is a non-receptor tyrosine kinase that normally relays the EPO signal into the cell. The acquired JAK2 V617F mutation makes the kinase constitutively active, so STAT5 fires without EPO:
- Normal: EPO binds its receptor → JAK2 phosphorylates STAT5 → controlled RBC production.
- PV: mutant JAK2 signals continuously → uncontrolled erythropoiesis (± megakaryocyte and granulocyte expansion).
- Feedback: the kidney senses the high Hct and suppresses EPO → serum EPO is low — the single best discriminator from secondary polycythemia.
Downstream, the rising RBC mass produces hyperviscosity (headache, dizziness, visual blurring, microvascular ischemia) and thrombosis (viscous blood plus abnormal, sticky platelets); extramedullary hematopoiesis and pooling cause splenomegaly; and increased basophils/mast cells release histamine, driving pruritus and peptic ulcer disease.
Clinical Presentation & Diagnosis
Symptoms stem from hyperviscosity, thrombosis, and histamine release; many patients are found incidentally on a routine CBC.
- Constitutional: fatigue, weakness, weight loss, night sweats.
- Hyperviscosity: headache, dizziness, tinnitus, blurred vision, "fullness" in the head.
- Skin (high-yield):
- Plethora — ruddy/red face, palms, and mucous membranes.
- Aquagenic pruritus — itching after a hot shower or bath.
- Erythromelalgia — burning pain + redness of fingers/toes, relieved by aspirin.
- Thrombosis (most dangerous): arterial (stroke, MI, TIA, digital ischemia) and venous (DVT, PE, and classically hepatic vein thrombosis).
- Abdomen: splenomegaly (~75%), hepatomegaly (~30%), peptic ulcer disease (↑ histamine).
- Bleeding paradox: despite high counts, platelet dysfunction → epistaxis and easy bruising.
| Important – فكرة سؤال | |
| A middle-aged or older patient with a plethoric (ruddy) face, itching after a hot shower (aquagenic pruritus), and splenomegaly = Polycythemia Vera until proven otherwise. First test is a CBC; confirm with low serum EPO + JAK2 V617F. | تذكر |
Suspect PV in any patient with unexplained Budd–Chiari syndrome (hepatic venous outflow obstruction) or other splanchnic-vein thrombosis. See the Budd–Chiari syndrome overview for the etiologies, presentation, and the role of JAK2 testing.
| فخ امتحاني – Budd–Chiari | |
| أي مريض يعاني من تخثر الوريد الكبدي (Budd–Chiari) بدون سبب واضح، يجب اعتباره مصاباً بـ Polycythemia Vera حتى يثبت العكس. | ملاحظة |
Diagnostic pathway (CBC → EPO/JAK2 → marrow)
- CBC + smear: ↑ Hb/Hct, often ↑ WBC and platelets (panmyelosis); normochromic normocytic RBCs (microcytic if iron-deficient from bleeding/phlebotomy).
- Serum EPO: low in PV — the best discriminator from secondary polycythemia (normal/high EPO).
- JAK2 mutation: V617F (~95%); if negative, test exon 12 (~3%).
- Bone marrow biopsy: hypercellular, panmyelosis, clustered atypical megakaryocytes, absent iron stores.
- Supporting clues: ↑ uric acid (gout), ↑ LDH, ↑ B12, ↑ leukocyte alkaline phosphatase (LAP) — high LAP helps separate PV from CML (low LAP).
| WHO 2016 Diagnostic Criteria for Polycythemia Vera: Diagnosis = all 3 major criteria, OR the first 2 major criteria + the minor criterion | |
| Major | All three required (criterion 2 may be waived if Hb/Hct markedly elevated) |
| 1. ↑ Hb / Hct | Hb >16.5 g/dL (men) or >16.0 g/dL (women); OR Hct >49% (men) or >48% (women) |
| 2. Bone marrow | Hypercellular for age with trilineage growth (panmyelosis): erythroid, granulocytic, and megakaryocytic proliferation |
| 3. JAK2 mutation | JAK2 V617F (~95%) or JAK2 exon 12 mutation (~3%) |
| Minor | Substitutes for major criterion 2 |
| Serum EPO | Subnormal (low) serum erythropoietin level |
Differential diagnosis
The core task is separating clonal PV (low EPO, JAK2⁺) from secondary (high EPO) and relative (normal RBC mass) polycythemia. See the primary vs secondary polycythemia differential for the full EPO-based work-up.
| Polycythemia Vera vs Secondary vs Relative Polycythemia | |||
|---|---|---|---|
| Feature | PV (Primary) | Secondary | Relative (Apparent) |
| RBC mass | ↑↑ | ↑ | Normal |
| Plasma volume | Normal / ↑ | Normal | ↓ |
| Serum EPO | ↓ LOW | ↑ HIGH | Normal |
| O₂ saturation | Normal | ↓ (if hypoxia-driven) | Normal |
| JAK2 mutation | Positive (~95%) | Negative | Negative |
| Splenomegaly | Yes | No | No |
| Pruritus / plethora | Yes | No | No |
| Top causes | Clonal JAK2⁺ stem cell | Hypoxia (COPD, OSA, high altitude); EPO-secreting tumors (RCC, HCC) | Dehydration, diuretics (Gaisböck) |
Among the other MPNs: ET = isolated ↑↑ platelets with normal Hct; PMF = cytopenias, teardrop cells, "dry tap," massive splenomegaly; CML = ↑↑ WBC with left shift, Philadelphia chromosome (BCR-ABL), and low LAP. The chronic myeloproliferative disorders comparison lays out the defining mutation for each.
Management
Treatment goals: reduce thrombosis risk, control symptoms, and prevent progression. Risk-stratify first, because the universal therapy is the same for everyone and only cytoreduction is added for high-risk disease:
- Low risk: age < 60 AND no prior thrombosis.
- High risk: age ≥ 60 OR prior thrombosis.
Universal therapy — all patients
- Step 1: Therapeutic phlebotomy — remove 250–500 mL of blood as needed to keep Hct < 45% (both sexes). This is the single most important intervention.
- Step 2: Low-dose aspirin 81 mg PO daily — reduces arterial thrombosis and relieves erythromelalgia. Hold if active bleeding or extreme thrombocytosis (> 1,500 × 10⁹/L → acquired von Willebrand disease).
- Step 3: Aggressive cardiovascular risk control — smoking cessation; treat hypertension, diabetes, and hyperlipidemia.
| ملاحظة سريرية هامة – الحديد | |
| نقص الحديد الناتج عن سحب الدم المتكرر (phlebotomy) هو هدف علاجي مقصود للحد من إنتاج كريات الدم الحمراء، ولا يجوز إعطاء المريض مكملات الحديد. | ملاحظة |
Add for high-risk patients — cytoreduction
- Hydroxyurea — first-line cytoreductive agent (suppresses marrow); watch for cytopenias and skin ulcers.
- Interferon-α / pegylated IFN — preferred in pregnancy and younger patients.
- Ruxolitinib — a JAK1/2 inhibitor for hydroxyurea-resistant or -intolerant disease; also controls pruritus and splenomegaly.
- Supportive: allopurinol for hyperuricemia/gout; antihistamines or SSRIs for refractory pruritus.
| Important – فكرة سؤال | |
| Hydroxyurea is the first-line cytoreductive agent for high-risk PV. Avoid alkylating agents (chlorambucil, busulfan) and radioactive ³²P — they increase the risk of transformation to AML. | تذكر |
Complications & Prognosis
- Thrombosis — arterial (stroke, MI) and venous (DVT, PE, Budd–Chiari). Leading cause of death.
- Hemorrhage — GI bleeding, epistaxis (platelet dysfunction; acquired von Willebrand disease at very high platelet counts).
- Hyperuricemia → gout and uric acid stones (high cell turnover).
- Peptic ulcer disease — from increased histamine release.
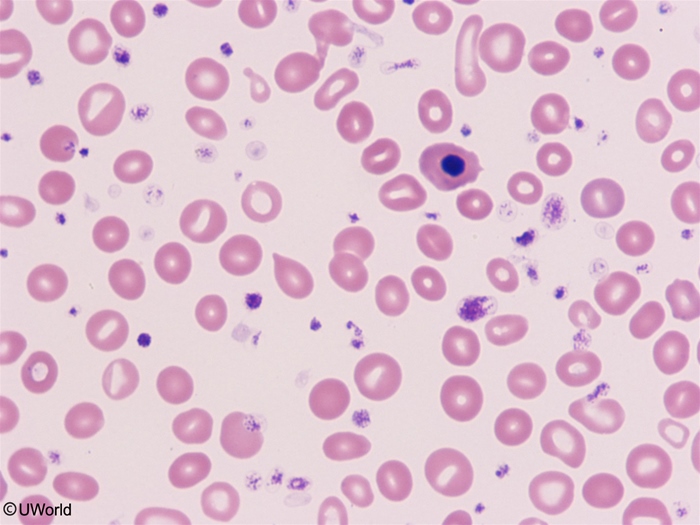
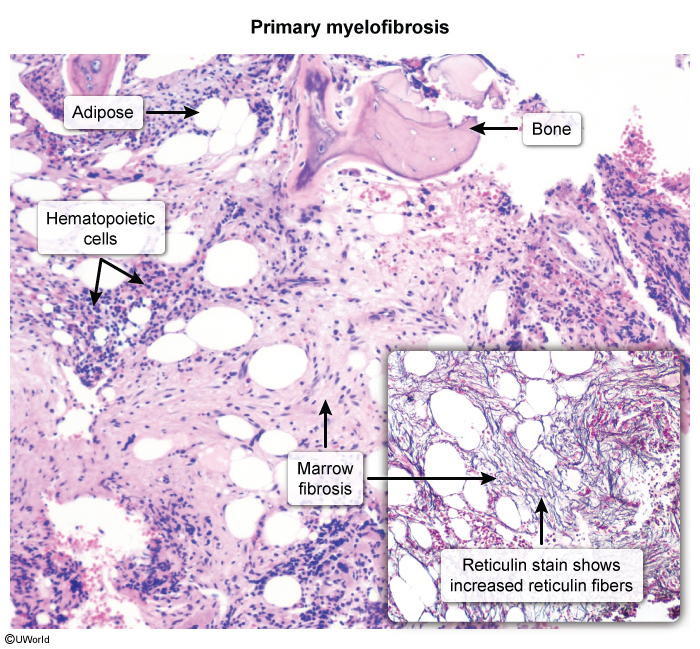
- Transformation — post-PV myelofibrosis ("spent phase," ~10–20% over years) — marrow fibrosis, falling Hb, worsening splenomegaly, teardrop cells (dacrocytes) on smear.
- Transformation to AML (~5–10%) — higher with alkylating-agent therapy.
Two smear/marrow clues mark progression to the spent phase: teardrop cells appear in the blood and the marrow shows reticulin fibrosis with a "dry tap." See the primary myelofibrosis overview for the pathogenesis and how post-PV fibrosis behaves.
Prognosis: with treatment, median survival exceeds 15–20 years. Untreated PV has a median survival of only ~18 months — driven largely by thrombotic events.
High-Yield Exam Pearls
The most testable points in one place:
- Mechanism: clonal JAK2 V617F → EPO-independent erythropoiesis → low serum EPO.
- Best discriminator: low EPO + JAK2⁺ = PV; high EPO = secondary; normal RBC mass with ↓ plasma volume = relative.
- Classic clues: aquagenic pruritus, facial plethora, erythromelalgia (aspirin-relieved), splenomegaly.
- Budd–Chiari in an otherwise unexplained patient = PV until proven otherwise.
- Treatment: phlebotomy to Hct < 45% + low-dose aspirin 81 mg for everyone; add hydroxyurea if age ≥ 60 or prior thrombosis.
- Phlebotomy-induced iron deficiency is intended — do NOT supplement iron.
- Thrombosis is the leading cause of death; long-term, watch for post-PV myelofibrosis and AML. Avoid alkylating agents.
| Mnemonic — "PV PHATBED" + treatment "PAH" | |
| Pruritus (aquagenic, after a hot shower) Hyperviscosity (headache, blurred vision, dizziness) Arterial & venous thrombosis (Budd–Chiari!) Transformation to myelofibrosis / AML Bleeding (acquired vWD at very high platelets) Erythromelalgia (burning hands/feet) Diagnosis: low EPO + JAK2 V617F Treatment = "PAH": Phlebotomy + Aspirin (all patients) + Hydroxyurea (high-risk) |
جملة تذكرية |
For a single-screen revision sheet, see the polycythemia vera summary covering manifestations, examination, labs, complications, and treatment.
احصل على التجربة الكاملة
اشترك للوصول لفيديوهات الشرح التفصيلي والبطاقات التعليمية التفاعلية وأسئلة الممارسة مع تتبع التقدم.