Summary
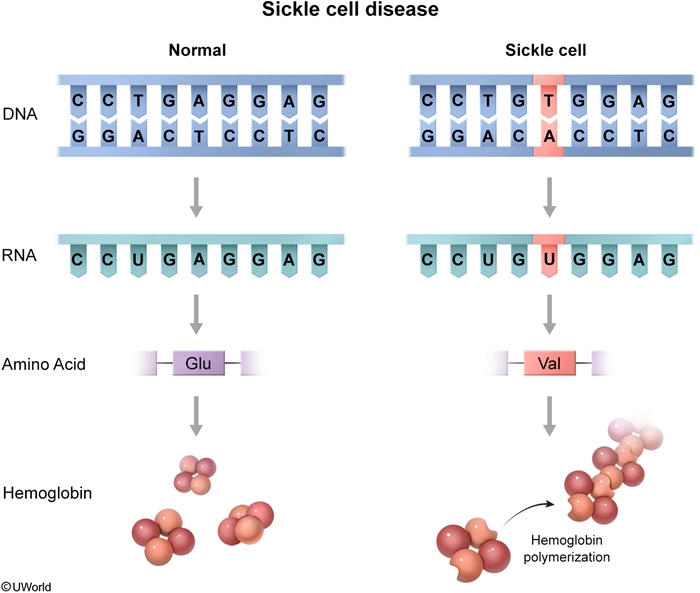
Sickle cell disease (SCD) is an autosomal recessive hemoglobinopathy caused by a point mutation in the β-globin gene that replaces glutamic acid with valine at position 6, producing abnormal hemoglobin S (HbS).
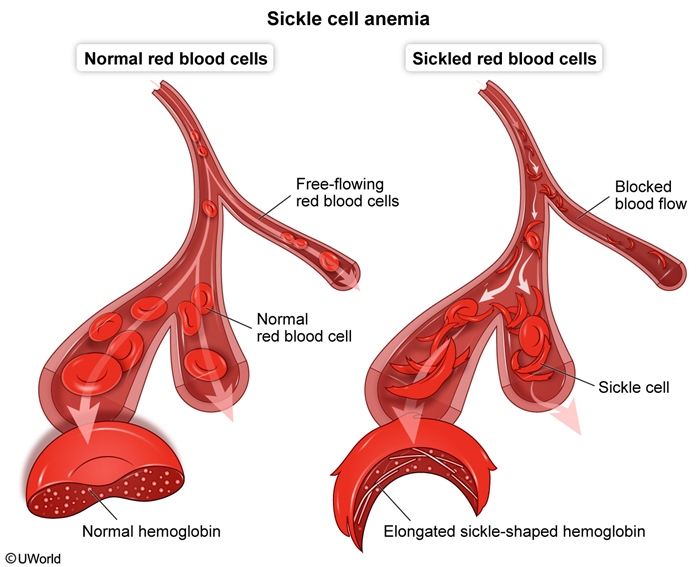
When deoxygenated, HbS polymerizes and distorts red blood cells into a rigid sickle shape. These cells block small vessels (vaso-occlusion) and are destroyed early (chronic hemolysis), causing the two main clinical pictures:
- Vaso-occlusive events: pain crises, dactylitis, acute chest syndrome, stroke, priapism, avascular necrosis.
- Chronic hemolytic anemia: jaundice, gallstones, aplastic crisis (parvovirus B19), splenic sequestration.
Diagnosis is confirmed by hemoglobin electrophoresis (HbSS pattern). Cornerstones of management are hydroxyurea (↑ HbF), vaccination and penicillin prophylaxis (autosplenectomy), folic acid, hydration, opioids for pain, and transfusion in severe events. Hematopoietic stem cell transplant is the only curative option.
Definition and Genetics
Sickle cell disease is a group of autosomal recessive disorders caused by mutations in the β-globin gene. The most common and severe form is sickle cell anemia (HbSS), in which the patient inherits two copies of the sickle mutation.
Genotypes you must know
- HbAA — Normal.
- HbAS — Sickle cell trait (heterozygous). Usually asymptomatic; carries one mutated allele.
- HbSS — Sickle cell anemia (homozygous). Most severe form.
- HbSC — Compound heterozygous (S + C). Milder course, more retinopathy and avascular necrosis.
- HbS/β-thalassemia — Severity depends on whether β⁰ (severe) or β⁺ (milder) thalassemia.
Normal adult hemoglobin is HbA (α₂β₂). Fetal hemoglobin HbF (α₂γ₂) does not contain β-chains, so it does not sickle — this is why newborns are protected for the first ~6 months and why hydroxyurea (which raises HbF) is therapeutic.
| Important – فكرة سؤال | |
|
The point mutation is GAG → GTG on the β-globin gene (chromosome 11), causing Glu → Val substitution at position 6 (E6V). Valine is hydrophobic — this is why HbS polymerizes when deoxygenated. |
تذكر |
Epidemiology
- Most common in people of African, Mediterranean (including parts of the Middle East), Indian, and Caribbean descent.
- The sickle allele is preserved in these populations because HbAS (trait) confers partial resistance to Plasmodium falciparum malaria — a classic example of heterozygote advantage.
- About 8% of African Americans carry sickle cell trait; ~1 in 400 has sickle cell anemia.
- Symptoms typically appear after 6 months of age, as HbF declines and HbS rises.
| Note – ملاحظة |
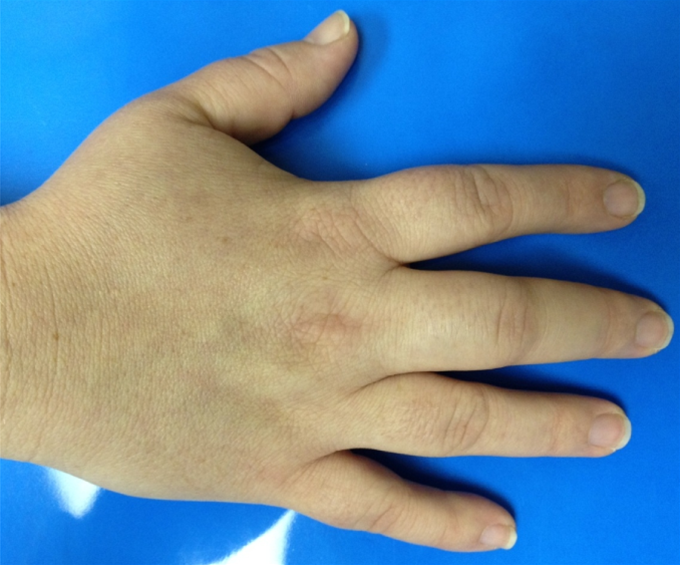
The first clinical clue in many infants is dactylitis (hand-foot syndrome) between 6 months and 4 years of age — painful, symmetric swelling of fingers/toes from microinfarction of small bones. |
Pathophysiology
The hydrophobic valine in HbS allows hemoglobin molecules to stick together when deoxygenated, forming long polymers inside the RBC. This produces the rigid, crescent-shaped sickle cell.
Two parallel mechanisms cause disease
- Vaso-occlusion — Rigid sickle cells get stuck in capillaries and post-capillary venules. Adhesion to endothelium, inflammation, and microthrombi cause ischemic tissue injury → pain, organ infarcts, dactylitis, stroke, acute chest syndrome.
- Chronic extravascular and intravascular hemolysis — Damaged sickle cells are destroyed by the spleen (extravascular) and also lyse in circulation (intravascular). Result: normocytic anemia, indirect hyperbilirubinemia, jaundice, pigment (bilirubin) gallstones, ↑ LDH, ↓ haptoglobin, and reticulocytosis.
Repeated splenic infarction in childhood leads to functional and then anatomic autosplenectomy, leaving patients vulnerable to encapsulated organisms (S. pneumoniae, H. influenzae, N. meningitidis, Salmonella).
| Mnemonic – Triggers of sickling | |
"HEDDA" — conditions that promote HbS polymerization:
أي شيء يقلل الأوكسجين أو يزيد الحموضة → يزيد تكوّن الخلايا المنجلية. |
جملة تذكرية |
Clinical Features
Symptoms begin after ~6 months of age when fetal hemoglobin (HbF) declines.
By organ system
- General: chronic fatigue, pallor, scleral icterus (from hemolysis), delayed growth and puberty.
- Bones/Joints:
- Dactylitis (hand-foot syndrome) — earliest manifestation in infants.
- Vaso-occlusive bone pain crises — most common reason for hospitalization.
- Avascular necrosis (AVN) of femoral/humeral head in adults.
- Salmonella osteomyelitis (classic association).
- Lungs: Acute chest syndrome (ACS) — leading cause of death in adults.
- CNS: stroke (ischemic in children, hemorrhagic in adults); silent infarcts.
- Spleen: early splenomegaly, then autosplenectomy → encapsulated organism infections; splenic sequestration crisis in young children.
- GU: priapism; renal papillary necrosis → painless hematuria, isosthenuria (loss of concentrating ability).
- Eyes: proliferative retinopathy (especially HbSC).
- Skin: chronic leg ulcers (medial malleolus).
- Hepatobiliary: pigment (bilirubin) gallstones → cholecystitis.
Diagnosis
Screening
- Newborn screening (universal in many countries) using hemoglobin electrophoresis or isoelectric focusing — detects all major hemoglobinopathies.
- Prenatal: chorionic villus sampling or amniocentesis if both parents are carriers.
Confirmatory test
Hemoglobin electrophoresis is the gold standard:
- HbAA (normal): ~97% HbA, 2–3% HbA₂.
- HbAS (trait): HbA (~55–60%) and HbS (~40–45%). Both bands present.
- HbSS (disease): only HbS and HbF; no HbA.
- HbSC: HbS + HbC; no HbA.
Routine labs in SCD
- CBC: normocytic, normochromic anemia (Hb ~6–9 g/dL); reticulocytosis.
- Peripheral smear: sickle cells, target cells, Howell–Jolly bodies (autosplenectomy), nucleated RBCs.
- Hemolysis markers: ↑ indirect bilirubin, ↑ LDH, ↓ haptoglobin, ↑ urine urobilinogen.
Peripheral blood smear in SCD Hemoglobin electrophoresis patterns Electrophoresis patterns in sickle cell syndromes
| Important – فكرة سؤال | |
|
The old sickle solubility test (Sickledex) is positive in both HbAS and HbSS — it cannot differentiate trait from disease. Always confirm with electrophoresis. |
تذكر |
Acute Complications (Crises)
Patients with SCD present to the emergency department with one of several distinct crises. Recognizing them is essential.
| Acute crises in sickle cell disease | ||||
|---|---|---|---|---|
| Crisis | Trigger / Cause | Key feature | Reticulocytes | Management |
| Vaso-occlusive (pain) crisis | Hypoxia, dehydration, cold, infection | Severe bone/back/chest pain | Normal or ↑ | IV fluids, O₂, opioids, NSAIDs |
| Acute chest syndrome | Infection, fat embolism, pulmonary infarct | Fever + new infiltrate + chest pain/hypoxia | Variable | O₂, IV fluids, ceftriaxone + azithromycin, exchange transfusion |
| Aplastic crisis | Parvovirus B19 (infects RBC precursors) | Sudden ↓ Hb, no jaundice change | ↓↓ (low) | Transfusion, supportive |
| Splenic sequestration | Trapping of blood in spleen (young children) | Rapid splenomegaly + shock | ↑ | Urgent transfusion, fluids; splenectomy if recurrent |
| Hyperhemolytic crisis | Infection, G6PD coexistence, drugs | ↓ Hb + ↑ jaundice + ↑ LDH | ↑↑ | Treat trigger, transfuse |
Acute Chest Syndrome (ACS)
The leading cause of death in adult SCD patients. Diagnostic triad:
- New pulmonary infiltrate on chest X-ray (involving ≥1 lung segment).
- Fever, chest pain, cough, dyspnea, or hypoxia.
- Often preceded by a pain crisis.
Empiric therapy: oxygen, IV fluids (caution: avoid overload), ceftriaxone + azithromycin (covers S. pneumoniae, Mycoplasma, Chlamydia), analgesia, incentive spirometry, and exchange transfusion if severe.
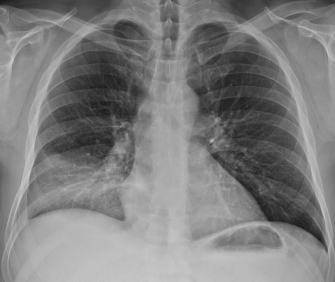 Acute chest syndrome — full criteria and management
Aplastic vs splenic sequestration crisis comparison
Acute chest syndrome — full criteria and management
Aplastic vs splenic sequestration crisis comparison
Chronic Complications
- Functional asplenia → infections with encapsulated organisms (S. pneumoniae sepsis, H. influenzae, N. meningitidis); Salmonella osteomyelitis.
- Avascular necrosis (AVN) — femoral head most common; hip pain, ↓ range of motion. MRI shows a serpiginous low-signal line.
- Stroke — peak risk age 2–10 years. Screen children with transcranial Doppler (TCD) ultrasound; chronic transfusion if abnormal.
- Pulmonary hypertension — late, poor prognosis.
- Renal: papillary necrosis (painless hematuria), isosthenuria (inability to concentrate urine), chronic kidney disease.
- Priapism — painful, prolonged erection from venous outflow obstruction; treat with hydration, analgesia, aspiration, intracavernous phenylephrine.
- Pigment (bilirubin) gallstones — chronic hemolysis releases unconjugated bilirubin.
- Chronic leg ulcers (medial malleolus).
- Iron overload from repeated transfusions → cardiomyopathy, liver/endocrine dysfunction; treat with chelators (deferasirox, deferoxamine).
- Proliferative retinopathy (especially HbSC).
- Delayed growth, delayed puberty.
| ملاحظة – Note |
في مرضى الخلايا المنجلية: التهاب العظم والنقي (osteomyelitis) سببه الأشهر هو Salmonella، لكن في عموم الناس يبقى Staphylococcus aureus هو الأكثر شيوعاً. هذه نقطة امتحان متكررة. |
Management
Chronic / long-term therapy
- Hydroxyurea — first-line disease-modifying drug. ↑ HbF, ↓ frequency of pain crises and ACS, ↓ transfusion need, ↓ mortality. Monitor CBC (myelosuppression).
- Folic acid daily — supports chronic erythropoiesis.
- Penicillin V prophylaxis — from 2 months until at least 5 years of age.
- Vaccinations: pneumococcal (PCV13 + PPSV23), meningococcal, H. influenzae type b, annual influenza, hepatitis B.
- Transcranial Doppler screening yearly ages 2–16 to predict stroke risk.
- Chronic transfusions for stroke prevention or recurrent severe complications; pair with iron chelation.
- Newer agents: L-glutamine, crizanlizumab (P-selectin inhibitor), voxelotor (HbS polymerization inhibitor).
- Hematopoietic stem cell transplant — the only curative option (HLA-matched sibling donor); gene therapy is emerging.
Acute pain crisis
- Hydration — IV normal saline (avoid overhydration → pulmonary edema/ACS).
- Oxygen if hypoxic.
- Analgesia — NSAIDs ± opioids (often morphine); do not under-dose.
- Treat any trigger — infection, dehydration, hypoxia.
- Transfusion — simple for severe anemia; exchange transfusion for ACS, stroke, multi-organ failure, priapism not resolving.
| Important – فكرة سؤال | |
Hydroxyurea works by raising fetal hemoglobin (HbF). HbF (α₂γ₂) lacks β-chains, so it cannot sickle. Side effect to remember: myelosuppression. الهيدروكسي يوريا ترفع الهيموجلوبين الجنيني (HbF)، وهو الذي يحمي من الانجلال. الأثر الجانبي الرئيسي: تثبيط نخاع العظم. |
تذكر |
Prevention and Routine Care
Because functional asplenia begins early, preventive care saves lives. Memorize this short checklist:
- Penicillin V prophylaxis — start at 2 months; continue to at least age 5.
- Vaccinations — pneumococcal (PCV + PPSV23), meningococcal (MCV + MenB), Hib, influenza yearly, hepatitis B.
- Folic acid — daily lifelong.
- Transcranial Doppler — yearly from age 2–16; if abnormal → chronic transfusion.
- Annual eye exam — screen for proliferative retinopathy (especially HbSC).
- Hydroxyurea — consider in all children ≥9 months and adults with HbSS.
- Education — avoid dehydration, cold, high altitude, strenuous exertion; recognize signs of crisis early.
- Fever in an SCD child = emergency — give empiric ceftriaxone after blood cultures (high risk of pneumococcal sepsis).
Sickle Cell Trait (HbAS)
Heterozygous carriers are typically asymptomatic and have normal lifespan, normal Hb, and normal smear. However, sickling can occur under extreme hypoxia or osmotic stress.
Complications you should still know
- Painless hematuria — from renal papillary microinfarction (renal medulla has low O₂ and high osmolarity → sickling).
- Isosthenuria / hyposthenuria — impaired urine concentration.
- Renal medullary carcinoma — rare but classic exam association with HbAS.
- Splenic infarct at high altitude.
- Exertional rhabdomyolysis / sudden death in extreme exercise (e.g., military training, athletes).
- Increased UTI risk in pregnancy.
Electrophoresis in trait: HbA ~55–60%, HbS ~40–45% (HbA > HbS because of unequal affinity of the chains).
Sickle cell trait — features, labs, and complicationsMnemonics
| Mnemonic – SICKLE | |
Splenic sequestration / autosplenectomy Infection (encapsulated, Salmonella osteomyelitis) Crisis — vaso-occlusive pain, ACS Kidney — papillary necrosis, isosthenuria Liver/biliary — pigment gallstones Erection (priapism), Eye (retinopathy), Extremities (dactylitis, AVN, ulcers) |
جملة تذكرية |
| Mnemonic – HEDDA | |
Triggers of sickling: Hypoxia · Exercise · Dehydration · Decreased pH (acidosis) · Altitude / cold |
جملة تذكرية |
| Mnemonic – Encapsulated organisms | |
"Some Killers Have Pretty Nice Capsules" Streptococcus pneumoniae · Klebsiella · Haemophilus influenzae · Pseudomonas · Neisseria meningitidis · Cryptococcus · (+ Salmonella, E. coli, group B strep) هؤلاء هم الذين يجب الحذر منهم في مرضى الخلايا المنجلية بسبب فقدان وظيفة الطحال. |
جملة تذكرية |
Key Points for Exams – نقاط مهمة للامتحانات
- Mutation: Glu → Val at position 6 of β-globin (GAG → GTG), chromosome 11. Autosomal recessive.
- HbF protects — symptoms start after 6 months. Hydroxyurea ↑ HbF.
- Dactylitis is often the first presentation in infants.
- Aplastic crisis = Parvovirus B19 → ↓ reticulocytes, ↓ Hb.
- Splenic sequestration → rapidly enlarging spleen + shock in a young child; reticulocytes ↑.
- Acute chest syndrome = new infiltrate + fever/hypoxia/chest pain → leading cause of adult death. Treat with O₂, fluids, ceftriaxone + azithromycin, ± exchange transfusion.
- Osteomyelitis: Salmonella in SCD (Staph aureus in general population).
- Functional asplenia → encapsulated organism sepsis. Penicillin prophylaxis + full vaccines are mandatory.
- Smear: sickle cells + Howell-Jolly bodies (autosplenectomy) + target cells.
- Diagnosis = hemoglobin electrophoresis (HbSS has only S and F; no HbA).
- Sickle trait: painless hematuria, isosthenuria, renal medullary carcinoma, splenic infarct at altitude, exertional rhabdomyolysis.
- Cure = HSCT; disease-modifier = hydroxyurea.
Sickle cell disease — comprehensive reference table
احصل على التجربة الكاملة
اشترك للوصول لفيديوهات الشرح التفصيلي والبطاقات التعليمية التفاعلية وأسئلة الممارسة مع تتبع التقدم.