Summary
Wilson disease (hepatolenticular degeneration) is a rare autosomal recessive disorder caused by mutations in the ATP7B gene, which regulates copper transport within hepatocytes. This leads to abnormal copper accumulation in the liver, brain, cornea, and other organs, resulting in hepatic, neurological, and psychiatric manifestations.
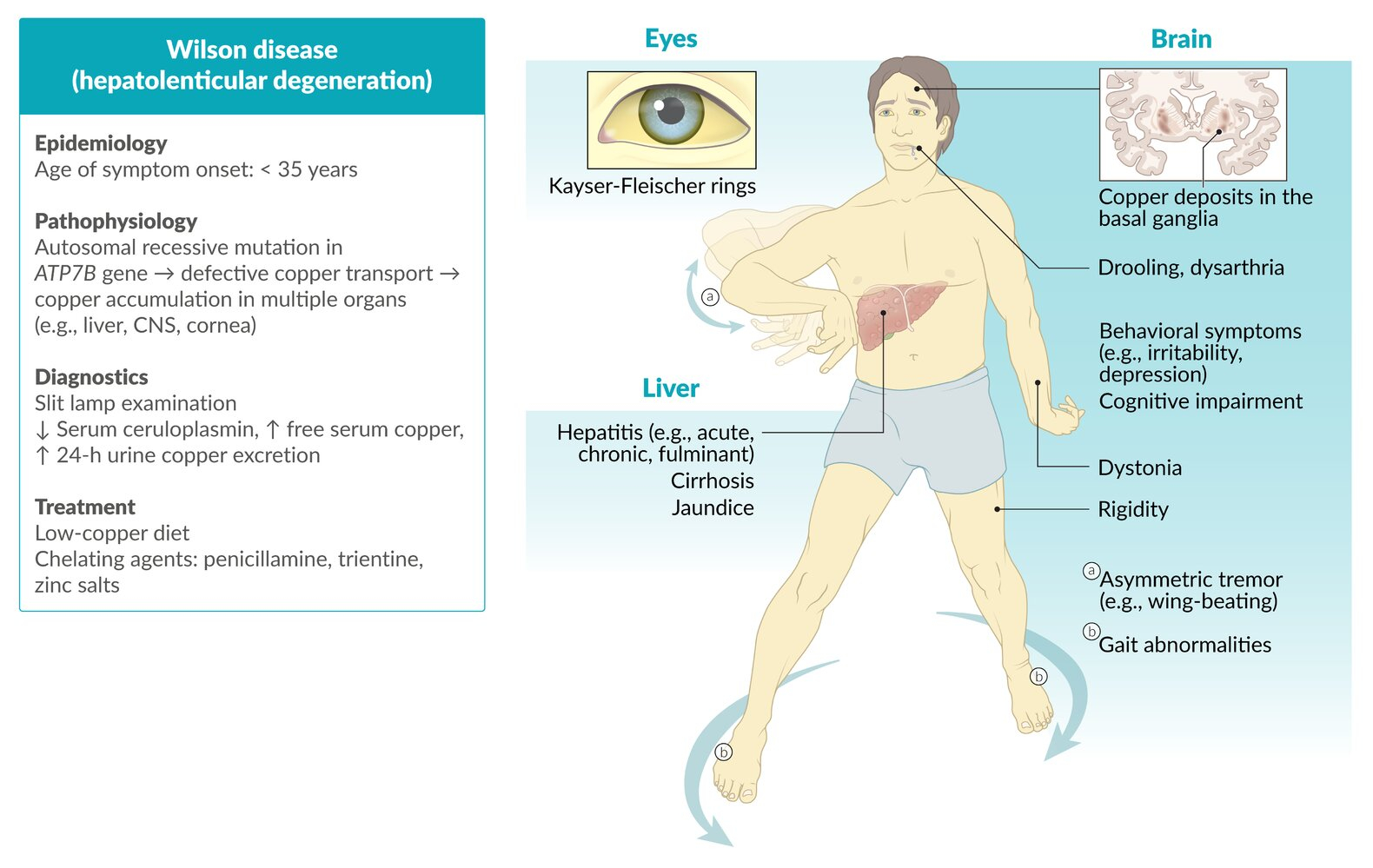
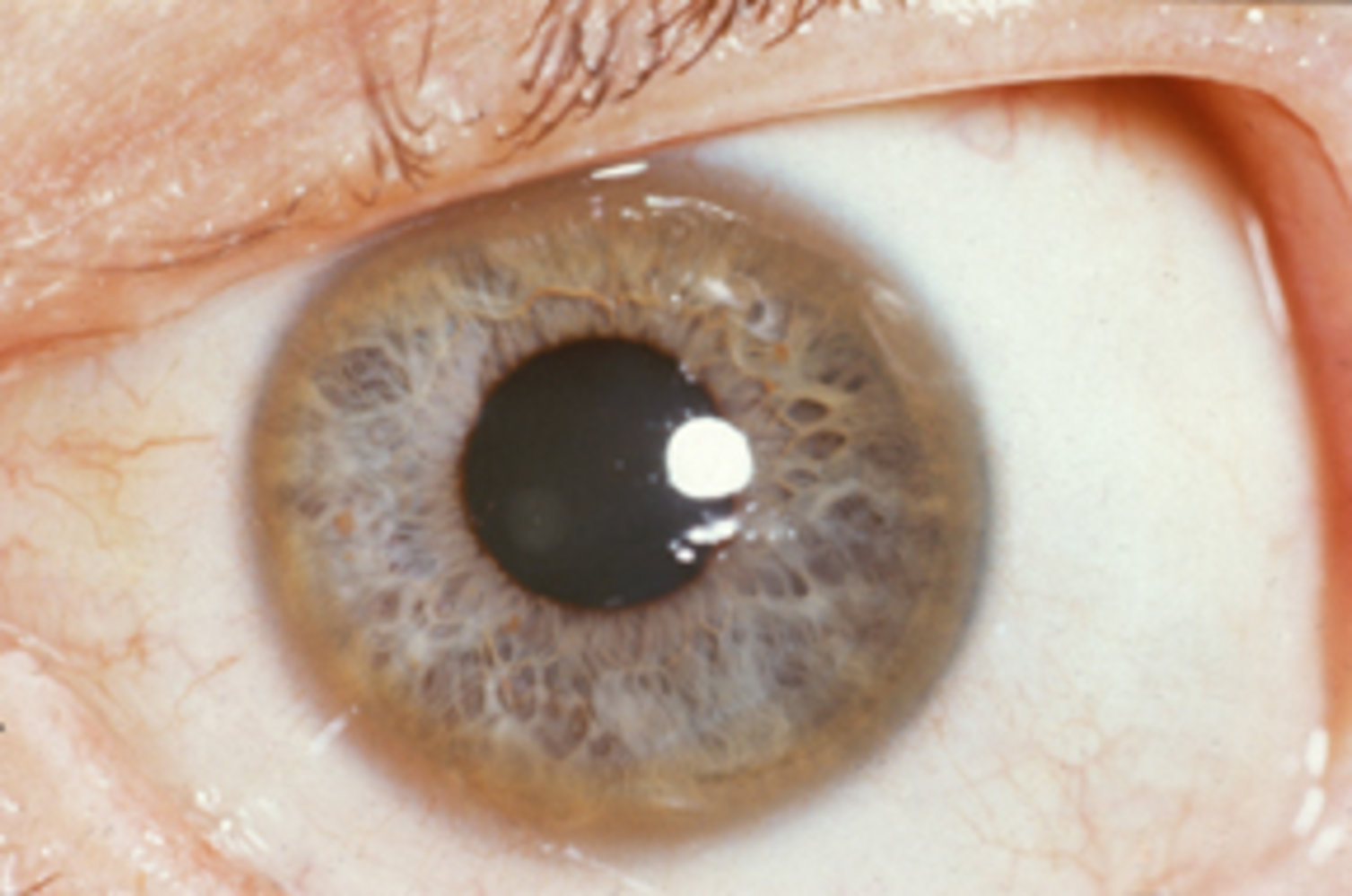
The disease typically presents between ages 5-35 years, with liver disease predominating in children and neuropsychiatric symptoms more common in older patients. Key diagnostic features include low serum ceruloplasmin, elevated urinary copper excretion, and Kayser-Fleischer rings on slit-lamp examination.
Treatment focuses on copper chelation therapy (primarily D-penicillamine or trientine) and preventing copper reaccumulation. Early diagnosis and treatment are crucial for preventing irreversible organ damage and improving long-term outcomes.
Definition and Overview
- Definition: Autosomal recessive disorder characterized by defective copper transport leading to toxic copper accumulation in tissues
- Gene mutation: ATP7B gene on chromosome 13 (encodes copper-transporting ATPase)
- Alternative name: Hepatolenticular degeneration
- Prevalence: 1 in 30,000 live births worldwide
- Age of onset: Typically 5-35 years (mean age at diagnosis: 13 years)
- Pattern:
- Children: predominantly hepatic manifestations
- Adolescents/Adults: neuropsychiatric symptoms more prominent
| Important – فكرة سؤال | |
| Wilson disease is caused by ATP7B gene mutation leading to defective copper transport. Remember: ATP7B = copper transporter protein. | تذكر |
Pathophysiology
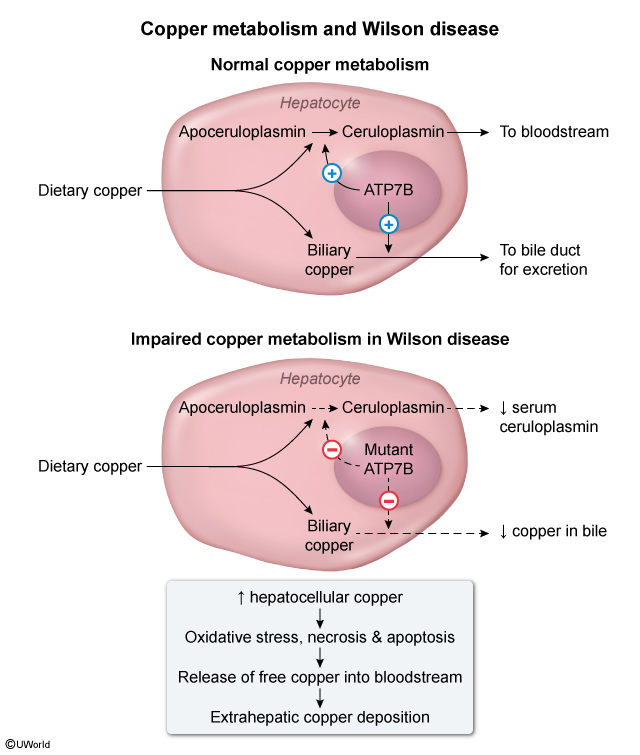
Normal Copper Metabolism:
- Dietary copper absorbed in stomach and duodenum → binds to albumin → transported to liver
- ATP7B protein in hepatocytes transfers excess copper to bile canaliculi
- Copper excreted via biliary system (majority of total body copper excretion)
- ATP7B also required for ceruloplasmin synthesis (copper-carrying protein)
Wilson Disease Pathophysiology:
- ATP7B mutation → impaired intracellular hepatocyte copper transport
- Decreased biliary copper excretion → hepatic copper accumulation
- Oxidative stress and hepatocyte apoptosis → liver dysfunction
- Copper release into bloodstream → extrahepatic tissue deposition:
- Brain (especially basal ganglia) → movement disorders
- Cornea → Kayser-Fleischer rings
- Kidney → renal dysfunction
- Decreased ceruloplasmin production → low serum ceruloplasmin (diagnostic marker)
ببساطة: عيب في نقل النحاس → تراكم النحاس في الكبد → تلف الكبد وتسرب النحاس → ترسب النحاس في الدماغ والعين والكلى
Clinical Features
| Clinical Manifestations of Wilson Disease | |
|---|---|
| Hepatic Manifestations |
|
| Neurological Manifestations |
|
| Psychiatric Manifestations |
|
| Ophthalmological |
|
| Other Manifestations |
|
| High-yield for Exams – فكرة سؤال | |
| Classic presentation: Young patient with unexplained liver disease + neuropsychiatric symptoms + Kayser-Fleischer rings. Think Wilson disease! | تذكر |
Diagnosis
Clinical Approach:
- Suspect in patients 5-35 years with unexplained liver disease, neuropsychiatric symptoms, or family history
- Screen first-degree relatives of diagnosed patients
Diagnostic Tests:
- Low serum ceruloplasmin (<20 mg/dL) → Most important initial test
- Normal: 20-40 mg/dL
- Low in 85% of Wilson disease patients
- Elevated 24-hour urinary copper excretion (>100 μg/day)
- Normal: <40 μg/day
- Wilson disease: usually >100 μg/day
- Slit-lamp examination: Kayser-Fleischer rings
- Pathognomonic when present
- Absence does not exclude diagnosis
Additional Studies:
- Liver biopsy: Elevated hepatic copper content (>250 μg/g dry weight)
- Genetic testing: ATP7B gene mutations (confirmatory)
- Brain MRI: Hyperintensities in basal ganglia, "face of giant panda" sign
- Laboratory findings:
- Elevated liver enzymes (AST, ALT)
- Low total serum copper
- Coombs-negative hemolytic anemia
| Note | |
| Diagnosis is confirmed by combination of: Low ceruloplasmin + High urinary copper + Clinical features. Single test may not be sufficient. | ملاحظة |
للتشخيص: انخفاض الـ ceruloplasmin + ارتفاع النحاس في البول + الأعراض السريرية. الفحص المؤكد هو خزعة الكبد أو الفحص الجيني.
Treatment
Goals of Treatment:
- Remove accumulated copper from tissues
- Prevent copper reaccumulation
- Lifelong treatment required
First-line Medications:
- D-Penicillamine (preferred)
- Copper chelating agent
- Dosage: 1-2 g/day in divided doses
- Take on empty stomach
- Co-administer with pyridoxine (vitamin B6)
- Side effects: rash, nephrotoxicity, bone marrow suppression
- Trientine
- Alternative copper chelator
- Used if penicillamine not tolerated
- Fewer side effects than penicillamine
Maintenance Therapy:
- Zinc supplementation
- Blocks intestinal copper absorption
- Used for asymptomatic patients or maintenance
- Dosage: 50 mg elemental zinc 2-3 times daily
- Take on empty stomach
Dietary Management:
- Low-copper diet (avoid high-copper foods)
- Avoid: organ meats, shellfish, nuts, chocolate, mushrooms
- Limit: liver, copper-containing water pipes
Surgical Treatment:
- Liver transplantation
- For fulminant liver failure
- Drug-resistant disease
- Advanced cirrhosis
- Curative treatment
| Treatment Pearl – فكرة سؤال | |
| First-line treatment: D-Penicillamine (copper chelator). Maintenance: Zinc supplementation. Emergency: Liver transplantation for fulminant failure. | تذكر |
Prognosis and Complications
Prognosis:
- Early diagnosis and treatment: Excellent prognosis
- Hepatic-only disease: Normal life expectancy with treatment
- Neuropsychiatric symptoms: May improve but some deficits may persist
- Untreated disease: Universally fatal
Complications:
- Cardiovascular disease → Leading cause of death
- Cardiomyopathy
- Arrhythmias
- Sudden cardiac death
- Progressive liver disease:
- Cirrhosis
- Portal hypertension
- Hepatocellular carcinoma (rare)
- Neurological complications:
- Progressive neurodegeneration
- Seizures
- Dementia
| Exam Focus – فكرة سؤال | |
| Main cause of death in Wilson disease: Cardiovascular complications. Early treatment prevents most complications and ensures normal life expectancy. | تذكر |
Differential Diagnosis
| Differential Diagnosis of Wilson Disease: Key Differentiating Features | |
|---|---|
| Condition | Key Differentiating Features |
| Hereditary Hemochromatosis |
|
| Autoimmune Hepatitis |
|
| α1-Antitrypsin Deficiency |
|
| Huntington Disease |
|
| Parkinson Disease |
|
Summary Table
| Wilson Disease - مرض ويلسون | |
|---|---|
| Definition |
|
| Clinical Features |
|
| Diagnosis |
|
| Treatment |
|
| Prognosis |
|
تذكر: مرض ويلسون = شاب + مرض كبد + أعراض عصبية + حلقات كايزر فليشر + انخفاض السيرولوبلازمين
احصل على التجربة الكاملة
اشترك للوصول لفيديوهات الشرح التفصيلي والبطاقات التعليمية التفاعلية وأسئلة الممارسة مع تتبع التقدم.